 The Green Lab Notebook (GLN) app has a little feature coming up in the next release: a mode for full screen display of an experiment, which consists of the reaction scheme and associated quantities. Continue reading
The Green Lab Notebook (GLN) app has a little feature coming up in the next release: a mode for full screen display of an experiment, which consists of the reaction scheme and associated quantities. Continue reading
cheminformatics
Tox21 toxicity measurements and PolyPharma
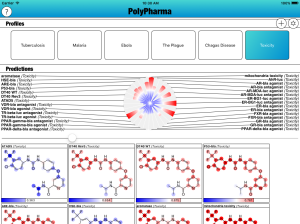 The PolyPharma app is currently getting some major new content added to it, namely a bunch of new models for toxicity. The models are derived from measurements made on the EPA’s Tox21 collection, recently published in nature, and released via PubChem. Continue reading
The PolyPharma app is currently getting some major new content added to it, namely a bunch of new models for toxicity. The models are derived from measurements made on the EPA’s Tox21 collection, recently published in nature, and released via PubChem. Continue reading
Adventures with KNIME: pipeline wrapping
 One of the items that has been on my to-do list for quite some time is to dig back into the KNIME universe, and interface it with the growing collection of cheminformatics algorithms that I have been assembling over the last few years. Given that my company’s software stack has its own workflow/pipelining infrastructure, but no user interface, it makes a certain amount of sense to look into connecting them together. Continue reading
One of the items that has been on my to-do list for quite some time is to dig back into the KNIME universe, and interface it with the growing collection of cheminformatics algorithms that I have been assembling over the last few years. Given that my company’s software stack has its own workflow/pipelining infrastructure, but no user interface, it makes a certain amount of sense to look into connecting them together. Continue reading
Panel of Bayesian screening for BIA 10-2474
The around town in drug discovery right this moment seems to be focused on BIA 10-2474, which my frequent collaborator Sean Ekins has weighed in on at the Collaborative Chemistry blog. In a spur of the moment effort to see if we could use some of our work-in-progress technologies to learn something about what’s going on, we ran it through a series of 1800 Bayesian models that we extracted from ChEMBL. For a detailed view, check out this link on molmatinf.com. The file is close to 20MB, so be patient if you’re on a slow connection. Continue reading
Composite Bayesian models: latest open source project
 My latest publication has just come out as early access in Journal of Chemical Information & Modeling, entitled “Open Source Bayesian Models: 3. Composite Models for Prediction of Binned Responses“. This is an extension of previous work on the Bayesian/fingerprint theme, and in the interim while waiting for peer review, we have some additional developments to share. Continue reading
My latest publication has just come out as early access in Journal of Chemical Information & Modeling, entitled “Open Source Bayesian Models: 3. Composite Models for Prediction of Binned Responses“. This is an extension of previous work on the Bayesian/fingerprint theme, and in the interim while waiting for peer review, we have some additional developments to share. Continue reading
Experiment editing in XMDS: dogfooding time
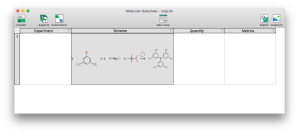 Progress is coming along nicely with the Experiment aspect editor for XMDS, which is essentially the desktop platform playing catchup with mobile and matching the functionality of the Green Lab Notebook (GLN) app. There is now an editor in place for the overall reaction scheme, which allows the components of a multistep reaction to be composed. This is a development milestone because it means that it’s time to switch to using it for data entry to find out what needs to be added or fixed most urgently, rather than implementing essential features that are obviously missing (cf. dogfooding = eating one’s own dogfood, in case anyone is unfamiliar with the term). Continue reading
Progress is coming along nicely with the Experiment aspect editor for XMDS, which is essentially the desktop platform playing catchup with mobile and matching the functionality of the Green Lab Notebook (GLN) app. There is now an editor in place for the overall reaction scheme, which allows the components of a multistep reaction to be composed. This is a development milestone because it means that it’s time to switch to using it for data entry to find out what needs to be added or fixed most urgently, rather than implementing essential features that are obviously missing (cf. dogfooding = eating one’s own dogfood, in case anyone is unfamiliar with the term). Continue reading
Visualisation of structure-activity models: fudging it with a widget
 One of the opinions (arguably of the educated variety) that I’ve been pushing for awhile now is the idea that when a model building or visualisation technique requires a user parameter in order to get the correct result, that is essentially an admission of partial failure. If the method really was so great, then it would be able to figure it out, because a parameter is an extra degree of freedom that the method has punted on. Now of course this is not a rule by any stretch of the imagination, and there are numerous exceptions, or grey areas between what’s a parameter and what’s an integral component of the source data. But sometimes a parameter really is just something that a method ought to know, but gives up and passes the burden on to the user – and that’s not necessarily a bad thing, as long as we admit it. Continue reading
One of the opinions (arguably of the educated variety) that I’ve been pushing for awhile now is the idea that when a model building or visualisation technique requires a user parameter in order to get the correct result, that is essentially an admission of partial failure. If the method really was so great, then it would be able to figure it out, because a parameter is an extra degree of freedom that the method has punted on. Now of course this is not a rule by any stretch of the imagination, and there are numerous exceptions, or grey areas between what’s a parameter and what’s an integral component of the source data. But sometimes a parameter really is just something that a method ought to know, but gives up and passes the burden on to the user – and that’s not necessarily a bad thing, as long as we admit it. Continue reading
Literature how-to for structure:activity Bayesian models (and open source)
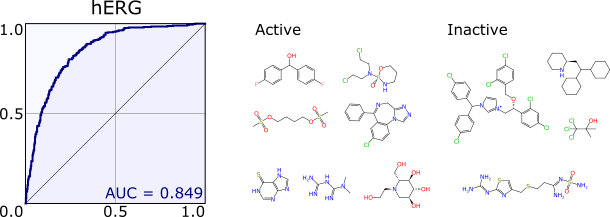 A two-pack of publications in Journal of Chemical Information and Modeling is now available: Bayesian the first, and Bayesian the second. Both papers are open access, so by all means go read them instead of this blog post. The first paper details the implementation of a variation of the classic naive Bayesian method that is suitable for use with structure-derived fingerprints such as ECFP6 and FCFP6. The text goes into some detail about how it is implemented, to the point of including pseudocode, which complements the fact that the source code is available as part of the Chemical Development Kit (CDK), conveniently and concisely coded up in a single source file. The intention is quite unashamedly to tell you everything you need to know to build the algorithm from scratch, should you be so inclined; and if not, to understand every little detail about how the open source software works. The second paper goes into some more detail about how to use this kind of (“Laplacian-modified”) Bayesian model, including a calibration method, and an extensive study carried out by extracting thousands of model-ready datasets from the ChEMBL database. Continue reading
A two-pack of publications in Journal of Chemical Information and Modeling is now available: Bayesian the first, and Bayesian the second. Both papers are open access, so by all means go read them instead of this blog post. The first paper details the implementation of a variation of the classic naive Bayesian method that is suitable for use with structure-derived fingerprints such as ECFP6 and FCFP6. The text goes into some detail about how it is implemented, to the point of including pseudocode, which complements the fact that the source code is available as part of the Chemical Development Kit (CDK), conveniently and concisely coded up in a single source file. The intention is quite unashamedly to tell you everything you need to know to build the algorithm from scratch, should you be so inclined; and if not, to understand every little detail about how the open source software works. The second paper goes into some more detail about how to use this kind of (“Laplacian-modified”) Bayesian model, including a calibration method, and an extensive study carried out by extracting thousands of model-ready datasets from the ChEMBL database. Continue reading
A rant about data quality: machines first, humans second…
 Recently one of my papers emerged through the publication system of Journal of Cheminformatics, entitled “Machines first, humans second: on the importance of algorithmic interpretation of open chemistry data“, co-authored with Antony Williams and Sean Ekins, and incorporated into the JC Bradley Memorial Issue. Spoiler alert: the paper is about how if you’re publishing open lab notebook data without adhering to rigorously defined standards for machine readability, then you’re mostly wasting your time, and arguably making the open data situation even worse than it already is. The tone of the article is a bit less polite than I normally try to be, so fair warning, but it’s all for a good cause.
Recently one of my papers emerged through the publication system of Journal of Cheminformatics, entitled “Machines first, humans second: on the importance of algorithmic interpretation of open chemistry data“, co-authored with Antony Williams and Sean Ekins, and incorporated into the JC Bradley Memorial Issue. Spoiler alert: the paper is about how if you’re publishing open lab notebook data without adhering to rigorously defined standards for machine readability, then you’re mostly wasting your time, and arguably making the open data situation even worse than it already is. The tone of the article is a bit less polite than I normally try to be, so fair warning, but it’s all for a good cause.
XMDS: progress toward structure sketcher
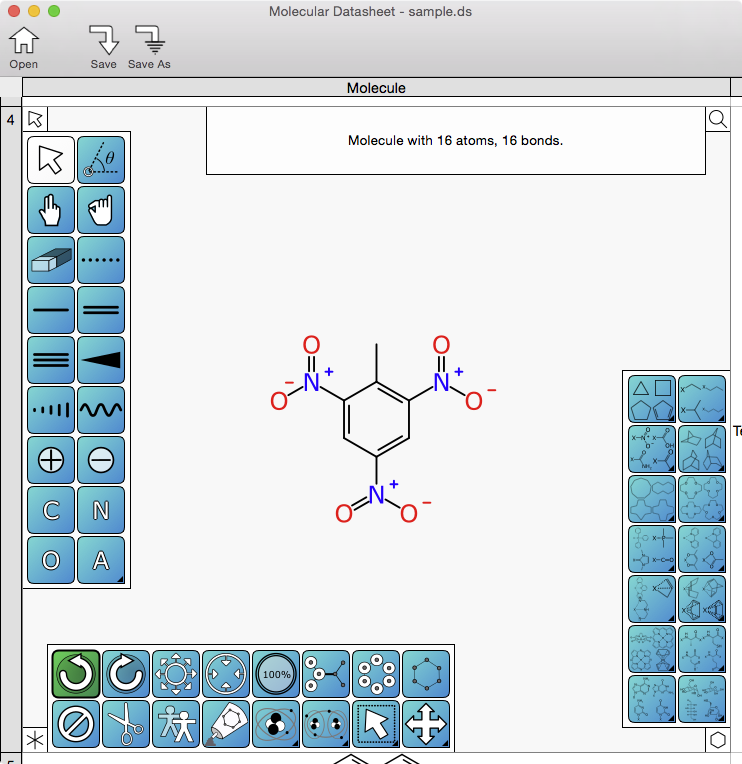 Since the last sneak preview, the skunkworks project “XMDS” – the Mac OS X desktop version of the Mobile Molecular DataSheet app – has gained enough functionality to make another screenshot, this time showing what the actual molecular drawing interface might look like once it’s done. Continue reading
Since the last sneak preview, the skunkworks project “XMDS” – the Mac OS X desktop version of the Mobile Molecular DataSheet app – has gained enough functionality to make another screenshot, this time showing what the actual molecular drawing interface might look like once it’s done. Continue reading
