11 September 2023
The BioAssay Express is being released as an open source project, under the Apache 2.0 license. The short description of this license is that it is permissive, and essentially the only restriction is acknowledgment.
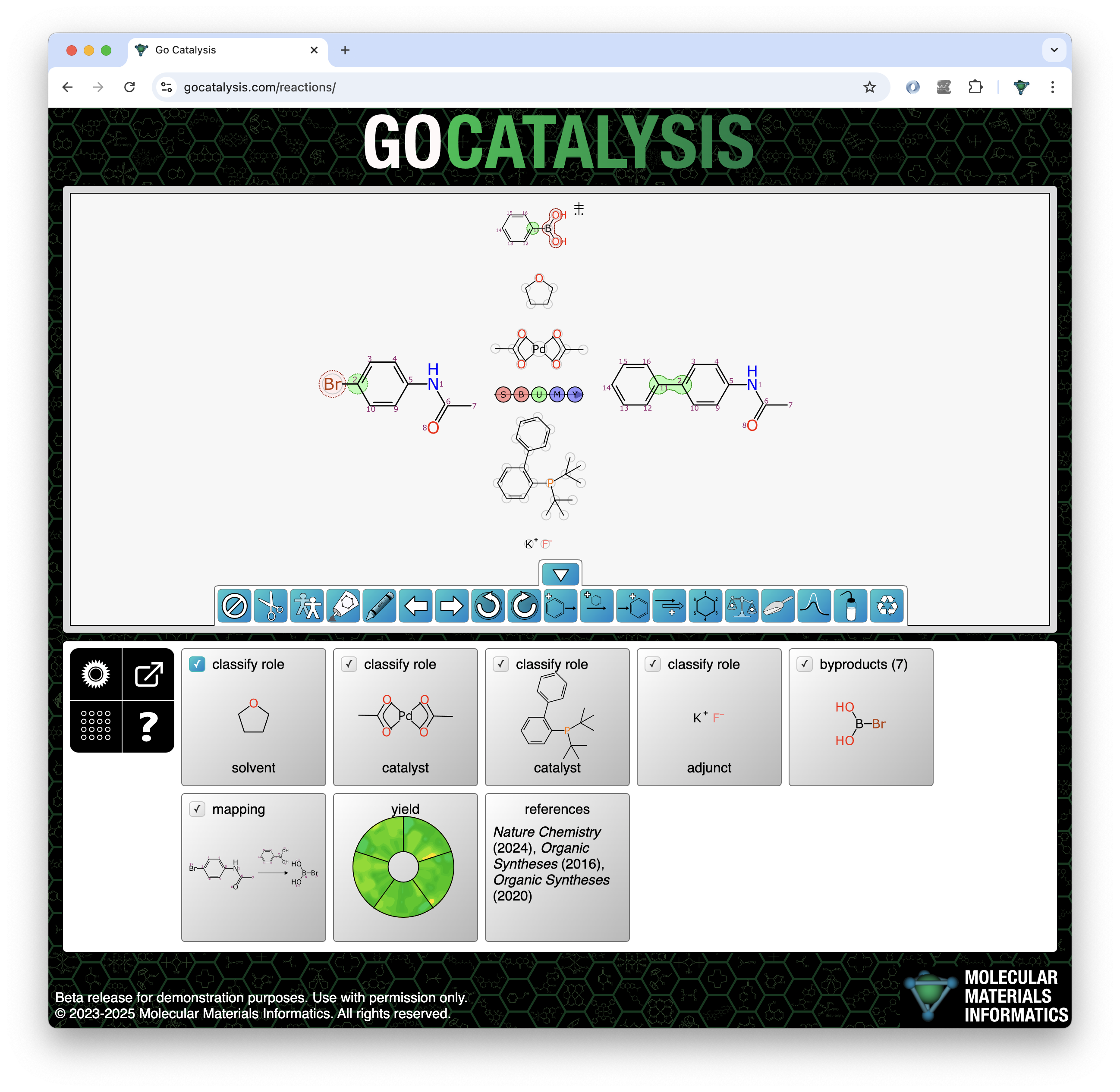
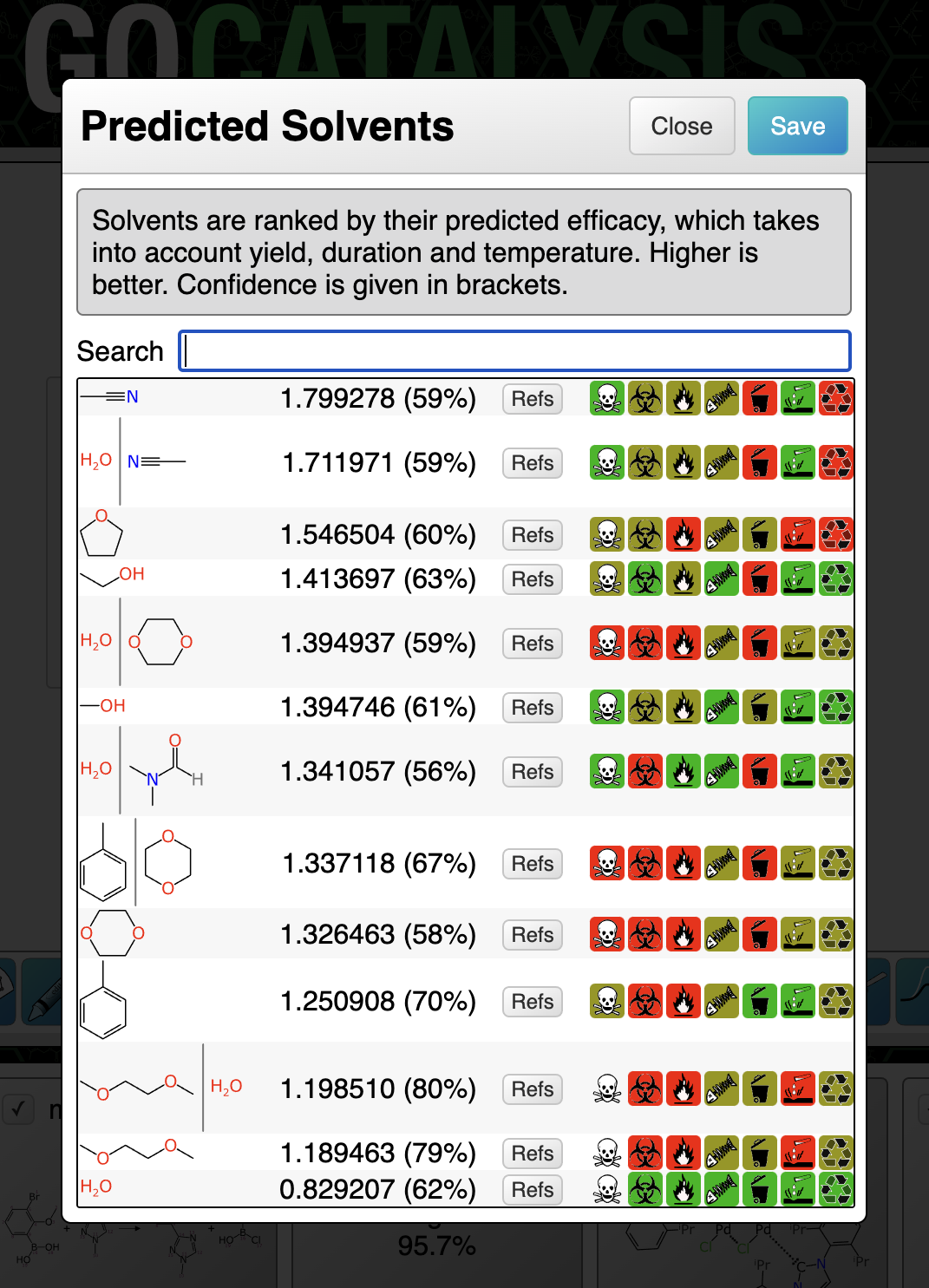
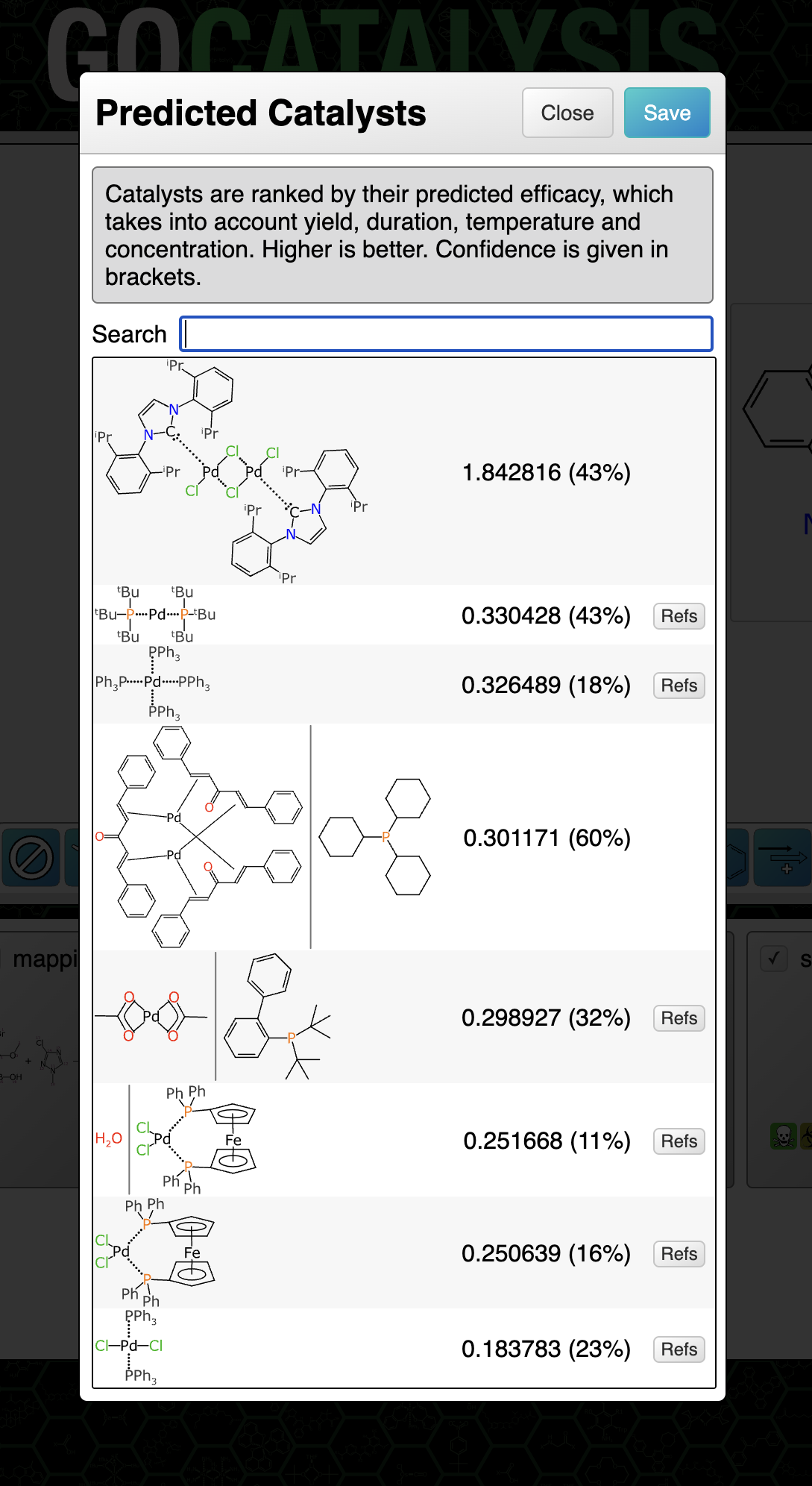
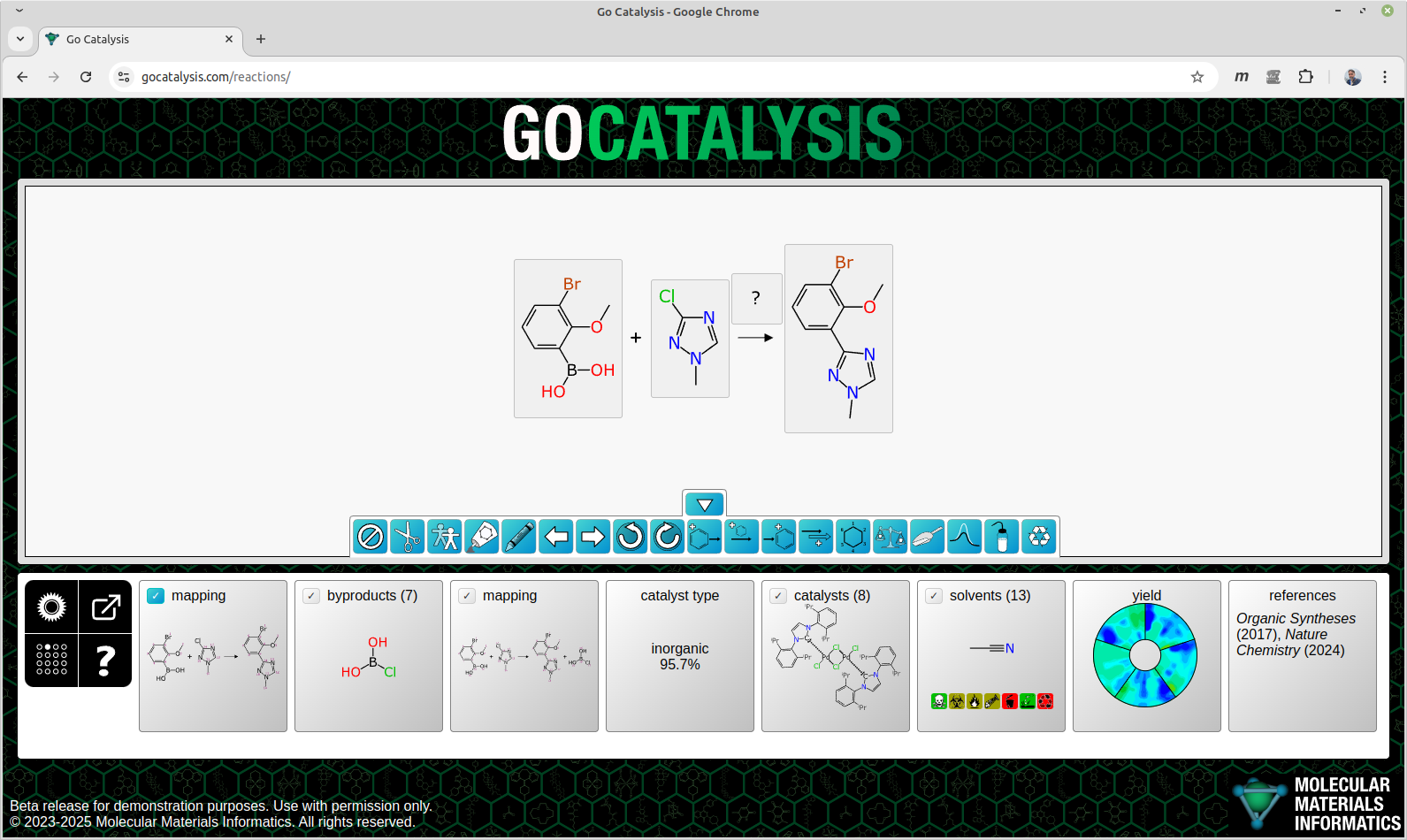
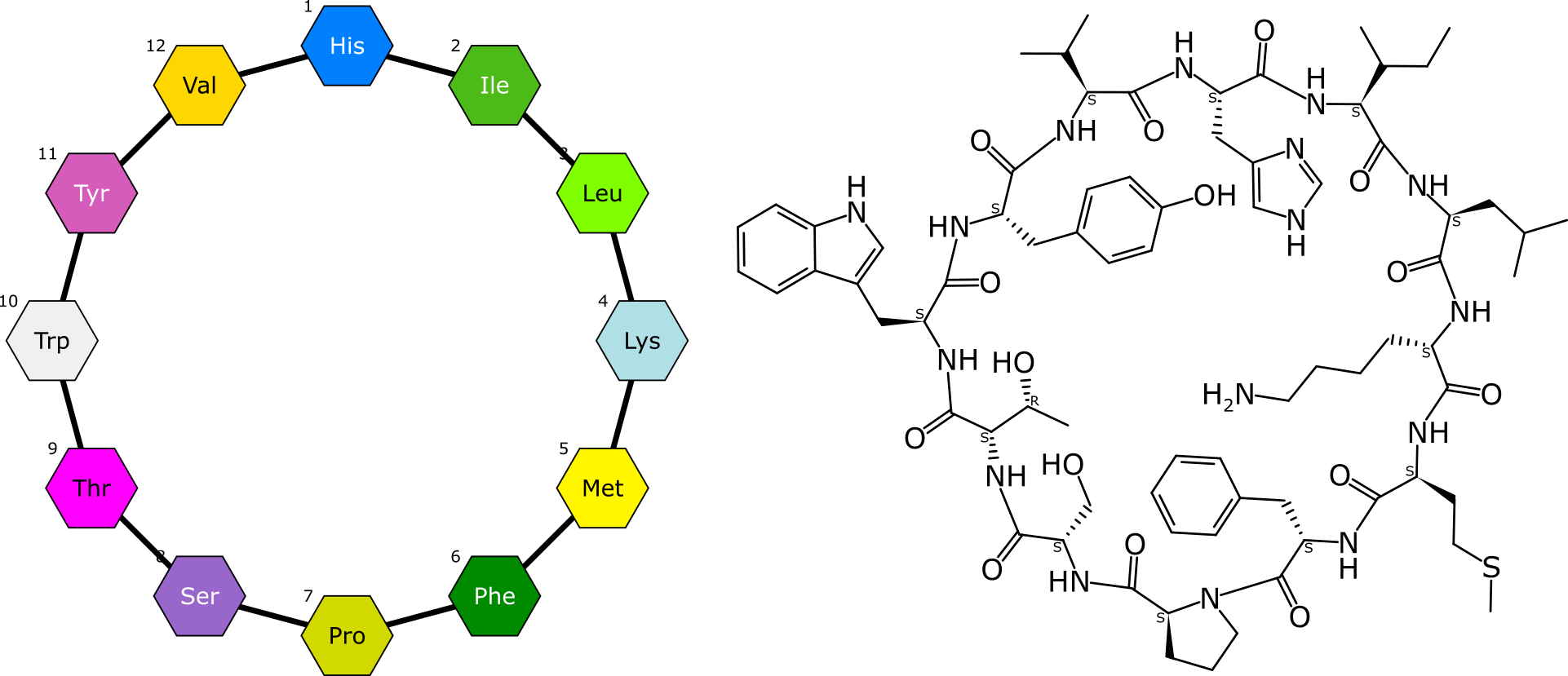
BioAssay Express is a grant-funded project to bring semantic web annotations to bioassay protocols, using vocabularies such as the BioAssay Ontology (BAO) to enrich descriptions that are primarily stored as text. Because of the universality of ontology terms, this means that annotated assays take on standardized meaning and can be processed by machines as effectively as they can be understood by scientists. This is a canonical example of the application of FAIR data principles (F = Findable, A = Accessible, I = Interoperable, R = Reusable).
Continue reading →