 Mobile apps for iOS have always been able to share files by a variety of different mechanisms, but many of these were limited in ways that were very detrimental to the user experience. The Green Lab Notebook app is now catching up to the new technology introduced with iOS 8: using the “document picker” interface to import and export files to document providers, which immediately makes it fully interoperable with iCloud, and file sharing services like Dropbox. Continue reading
Mobile apps for iOS have always been able to share files by a variety of different mechanisms, but many of these were limited in ways that were very detrimental to the user experience. The Green Lab Notebook app is now catching up to the new technology introduced with iOS 8: using the “document picker” interface to import and export files to document providers, which immediately makes it fully interoperable with iCloud, and file sharing services like Dropbox. Continue reading
Month: January 2015
Structure property calculation in apps: MMDS
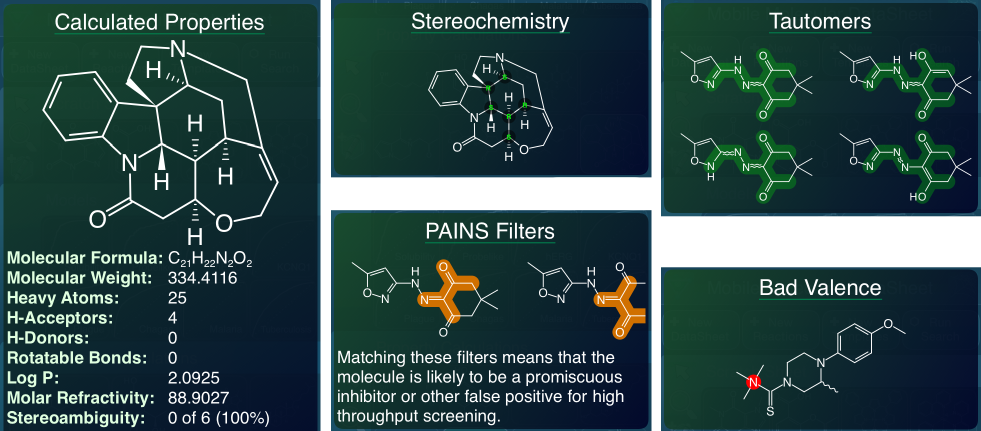 An important milestone in has been reached in the migration of complicated structure-based calculations to pure mobile. The latest version of MMDS (1.5.9) is now available on the AppStore, and allows visualisation of calculated properties for individual molecules, as well as calculating new columns for entire datasheets. Continue reading
An important milestone in has been reached in the migration of complicated structure-based calculations to pure mobile. The latest version of MMDS (1.5.9) is now available on the AppStore, and allows visualisation of calculated properties for individual molecules, as well as calculating new columns for entire datasheets. Continue reading
PAINS filters now on mobile, with MolPrime+
 One of the trends that you should expect to see more of from apps produced by Molecular Materials Informatics is a shift toward performing more advanced calculations internally on the mobile device, rather than calling out to a cloud service. One of the recent demonstrations was shown with the Approved Drugs app, which can now call up Bayesian models for various predictions. The next version of MolPrime+ that is awaiting review on the AppStore incorporates internal calculation of log P, and also brings the ability to identify PAINS filters for molecular structures.
One of the trends that you should expect to see more of from apps produced by Molecular Materials Informatics is a shift toward performing more advanced calculations internally on the mobile device, rather than calling out to a cloud service. One of the recent demonstrations was shown with the Approved Drugs app, which can now call up Bayesian models for various predictions. The next version of MolPrime+ that is awaiting review on the AppStore incorporates internal calculation of log P, and also brings the ability to identify PAINS filters for molecular structures.
