 The most recent addition to the OS X Molecular DataSheet (XMDS) desktop app is calculation of stereochemistry labels as-you-edit, using the Cahn-Ingold-Prelog (CIP) designations (R/S, Z/E). The labelling can be switched off with a menu option, but since most people use software with its factory settings, this is more or less equivalent to having it hardcoded permanently. Continue reading
The most recent addition to the OS X Molecular DataSheet (XMDS) desktop app is calculation of stereochemistry labels as-you-edit, using the Cahn-Ingold-Prelog (CIP) designations (R/S, Z/E). The labelling can be switched off with a menu option, but since most people use software with its factory settings, this is more or less equivalent to having it hardcoded permanently. Continue reading
Month: June 2015
Aspects and XMDS: higher order metadata for datasheets
 Recent progress on the OS X Molecular DataSheet (XMDS) app, which is currently in beta, has involved the inclusion of aspects into the user interface for editing datasheets. The core format that the XMDS desktop app, as well as all the rest of the products from Molecular Materials Informatics, is the datasheet, which is a tabular format made up of rows and columns: like a spreadsheet, except that the columns are strongly typed, so you can’t just abuse it and put in whatever you want wherever you want. The minimalistic table datastructure can be supplemented by any number of aspects, which are directives that impose higher order layers of interpretation, which are often composed of multiple columns (e.g. a chemical reaction which cobbles together multiple molecules for its reactants, reagents and products). One of the fun software engineering challenges has been to make these complex representations fit seemlessly into the grid-based “spreadsheet” editor upon which XMDS operates. Continue reading
Recent progress on the OS X Molecular DataSheet (XMDS) app, which is currently in beta, has involved the inclusion of aspects into the user interface for editing datasheets. The core format that the XMDS desktop app, as well as all the rest of the products from Molecular Materials Informatics, is the datasheet, which is a tabular format made up of rows and columns: like a spreadsheet, except that the columns are strongly typed, so you can’t just abuse it and put in whatever you want wherever you want. The minimalistic table datastructure can be supplemented by any number of aspects, which are directives that impose higher order layers of interpretation, which are often composed of multiple columns (e.g. a chemical reaction which cobbles together multiple molecules for its reactants, reagents and products). One of the fun software engineering challenges has been to make these complex representations fit seemlessly into the grid-based “spreadsheet” editor upon which XMDS operates. Continue reading
XMDS ready for beta phase
 The OS X Molecular DataSheet (XMDS) desktop app is now ready for beta testing. And by beta testing, I mean the minimum viable product is done and ready to be used for actual cheminformatics tasks. You can sign up anytime: all it takes is an email (info@molmatinf.com), a Mac with Yosemite-or-later (v10.10), and a Dropbox account. And you get to keep using the app for as long as you want, even after the beta testing programme is wrapped up. Continue reading
The OS X Molecular DataSheet (XMDS) desktop app is now ready for beta testing. And by beta testing, I mean the minimum viable product is done and ready to be used for actual cheminformatics tasks. You can sign up anytime: all it takes is an email (info@molmatinf.com), a Mac with Yosemite-or-later (v10.10), and a Dropbox account. And you get to keep using the app for as long as you want, even after the beta testing programme is wrapped up. Continue reading
Literature how-to for structure:activity Bayesian models (and open source)
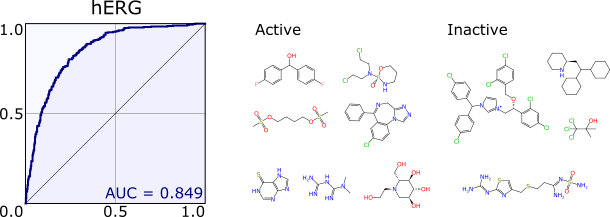 A two-pack of publications in Journal of Chemical Information and Modeling is now available: Bayesian the first, and Bayesian the second. Both papers are open access, so by all means go read them instead of this blog post. The first paper details the implementation of a variation of the classic naive Bayesian method that is suitable for use with structure-derived fingerprints such as ECFP6 and FCFP6. The text goes into some detail about how it is implemented, to the point of including pseudocode, which complements the fact that the source code is available as part of the Chemical Development Kit (CDK), conveniently and concisely coded up in a single source file. The intention is quite unashamedly to tell you everything you need to know to build the algorithm from scratch, should you be so inclined; and if not, to understand every little detail about how the open source software works. The second paper goes into some more detail about how to use this kind of (“Laplacian-modified”) Bayesian model, including a calibration method, and an extensive study carried out by extracting thousands of model-ready datasets from the ChEMBL database. Continue reading
A two-pack of publications in Journal of Chemical Information and Modeling is now available: Bayesian the first, and Bayesian the second. Both papers are open access, so by all means go read them instead of this blog post. The first paper details the implementation of a variation of the classic naive Bayesian method that is suitable for use with structure-derived fingerprints such as ECFP6 and FCFP6. The text goes into some detail about how it is implemented, to the point of including pseudocode, which complements the fact that the source code is available as part of the Chemical Development Kit (CDK), conveniently and concisely coded up in a single source file. The intention is quite unashamedly to tell you everything you need to know to build the algorithm from scratch, should you be so inclined; and if not, to understand every little detail about how the open source software works. The second paper goes into some more detail about how to use this kind of (“Laplacian-modified”) Bayesian model, including a calibration method, and an extensive study carried out by extracting thousands of model-ready datasets from the ChEMBL database. Continue reading
