 Further to the previous post that mentioned a new atom-mapping interface for the Mobile Molecular DataSheet (MMDS) app, the next step is an atom-to-atom pairing interface for reactions. A very short 30 second YouTube clip will show you how this works.
Further to the previous post that mentioned a new atom-mapping interface for the Mobile Molecular DataSheet (MMDS) app, the next step is an atom-to-atom pairing interface for reactions. A very short 30 second YouTube clip will show you how this works.
The interface style involves a very simple tap:tap sequence for pairing atoms together by selecting matched pairs from either side of the reaction arrow. While that’s a fast and quite convenient way to specify the relationship between reactant and product atoms, there is also an “automap” feature, which is demonstrated in the clip. Actually it works like more of an “auto-complete”, because it takes the user-provided matches as a starting point, and conservatively grows the set of selected atoms as far as possible until it runs out of options.
By conservative, it means that a mapping is only made if it is very obvious. For example, consider the partially matched-up reaction:

In this case there is just one unmatched atom, and the assignment between reactant and product is what would be known as, in the parlance of our times, a no-brainer.
The next example though presents a choice: the atom that is already assigned as number 1 is bonded to two unassigned carbon atoms that are undistinguishable from each other with a graph distance of one, on both sides of the reaction. There are a total of 4 (2×2) ways to assign the next atom mapping, and half of them are distinctly preferable, i.e. it would be better to assign atom number 3 so that the diagrams line up when they are laid on top of one another.
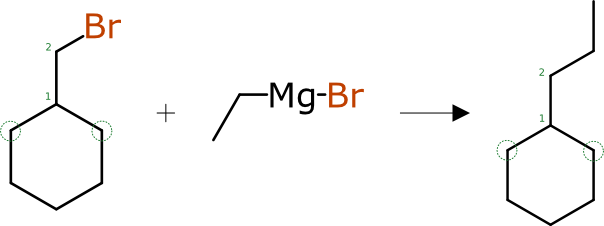
So for such cases, the algorithm takes into account the bond angles relative to the already-assigned atoms. In this way it is possible for the automation algorithm to assign all of the cyclohexane ring in an idealised way.
Other than a bit of trigonometry, the assignment algorithm is very simple: if it finds anything it can’t resolve as being immediately unambiguous, it won’t assign it. That means that with well drawn structures, most of the time the user just needs to assign several atoms in the active transform region, and most of the rest can be filled in automatically.
There is a strategic reason why this user interface has been constructed now, namely that I require the ability to create reaction atom mapping content for use with new products and features that are under development. Needless to say more details will be posted on this blog at a later date.

My experience has been that the novelty of adding reaction mapping among chemists wanes very rapidly, would it be possible to have the automatic mapping first and then the chemists needs to just click OK? Or make hopefully minor edits.
Perhaps the algorithm could “learn” from previous reactions?
What’s the use case that you have in mind? My original need is to be able to specify reference transforms and with 100% success, and get it done quickly with minimum pain. Specific use cases could be made much more convenient, with a bit of prior knowledge about the workflow…