 The TB Mobile app for iOS has just received a major new update on the iTunes AppStore. Version 2 of the app brings some fairly powerful cheminformatics functionality to this free mobile app for tuberculosis research. Produced by Collaborative Drug Discovery, built by Molecular Materials Informatics, and organised by Sean Ekins, this app is pushing the boundaries of real science on mobile devices in several important ways.
The TB Mobile app for iOS has just received a major new update on the iTunes AppStore. Version 2 of the app brings some fairly powerful cheminformatics functionality to this free mobile app for tuberculosis research. Produced by Collaborative Drug Discovery, built by Molecular Materials Informatics, and organised by Sean Ekins, this app is pushing the boundaries of real science on mobile devices in several important ways.
Relative to version 1, the first thing you will notice is that there has been some re-theming to go with migrating to iOS 7: the icons have a more platform-specific aesthetic, and the status bar is blended into the background. Besides window-dressing, the app has several key pieces of technology behind the scenes, and several major new interactive features. The previous version of the app had two major things going for it: (1) an expert-curated list of structures and associated target information for the tuberculosis mycobacterium, hitherto not conveniently available; and (2) presenting the data in the form of an app, that anyone can download for free. While it was possible to organise and select parts of the data (e.g. by name, target, simple molecular similarity) and export it in several forms, the cheminformatics functionality was fairly limited.
One of the first major extensions is the inclusion of a “personal stash” of molecular structures, which can be viewed and edited, by drawing structures with the built-in structure editor, or by importing from other apps:

Besides being a convenient reference collection, the personal stash gets interesting when you hit the prediction button. Each of the 90 or so tuberculosis targets (from ~800 curated compounds) is analysed with respect to each user supplied compound. A Bayesian model is used (with a new implementation of ECFP6-style fingerprints: more about this later). Likelihood of being active against any given target is rated from 0 to 1, and anything interesting is shown alongside the compound as a partially-filled bar, which you can see in the snapshot above.
The next major new feature is an interactive visual clustering view:
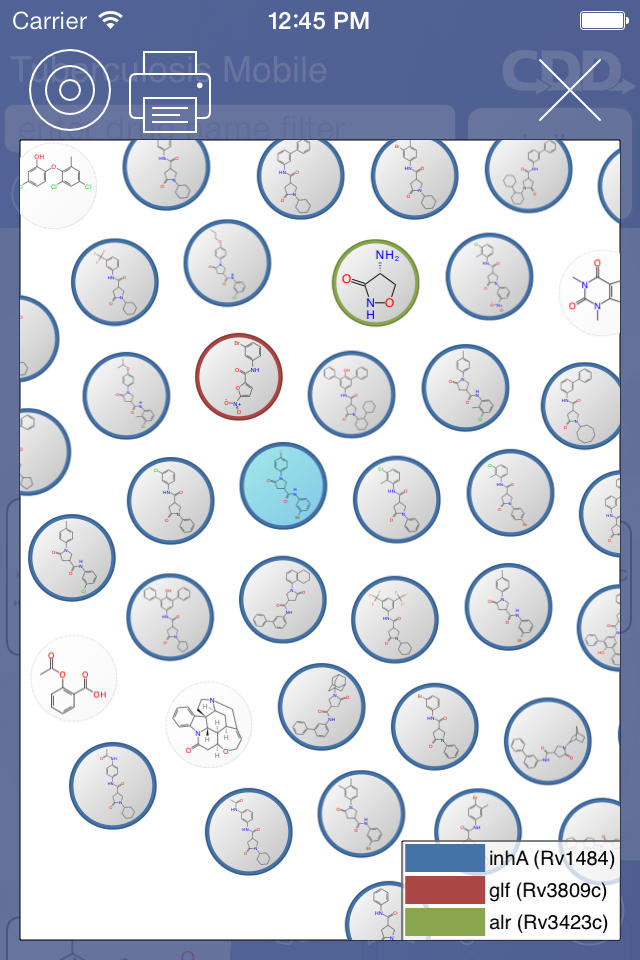
The way this works is that a reference compound is used as the central point (selected depending on where the view was invoked from), and similar compounds are arranged around them, in terms of most-similar-first. Because similarity is an n-dimensional property, and computer screens have 2 dimensions, the embedding has a certain amount of arbitrary bias, and so this view is actually interactive: you can tap on a molecule to select it, use your finger to “push” it elsewhere, then watch the rest of the molecules relax into new positions, based on invisible tethers determined by similarity. You can also delete molecules you don’t want in the view. The scientific motivation behind this is that you may want to cherrypick certain compounds in order to conceptualise how they related together in terms of structural similarity; but the motivation does include a significant amount of novelty value, too!
As well as structures, known targets can be highlighted by selecting from a list, which are colour-coded. And also, structures drawn into your personal stash are included in the cluster, to provide an idea of how they relate to known compounds in terms of structural similarity. And there is also an option to create a PDF file, and export or print it, in case you want to show the cluster to anyone else.
There are a number of other features, but that’s a start. The ECFP6-style fingerprints are used for all methods for determining structural similarity, including the similarity sorting feature accessible from the main list, which replaces the fairly crude method previously used. There are also quite a few performance optimisations and precalculated quantities, so the app should be more responsive in several important areas.
If you have any interest in tuberculosis research, and you have an iThing, check it out. It’s free, and it’s on the AppStore now! And thank Collaborative Drug Discovery for making this happen.
