There has been a lot of work, many products, and plenty said on the topic of converting chemical structures from one format to another, and a number of well used and highly regarded packages (e.g. OpenBabel) that specialise in providing this capability. Less well discussed, it would seem, is that when it comes to connection-table formats used to encode chemists’ sketches of molecules, there are no software packages and no two formats for which a round trip conversion can be reliably carried out without losing information, even if the chemical entities being expressed fall within the applicable domain of both formats.
And this is not just because most available cheminformatics software is bad (although this is certainly the case), but mostly because it is actually by definition impossible, due to the way the datastructures have been designed.
To illustrate the point, I want to use six format examples, and consider pairwise interactions between them, and how often format conversion is viable:
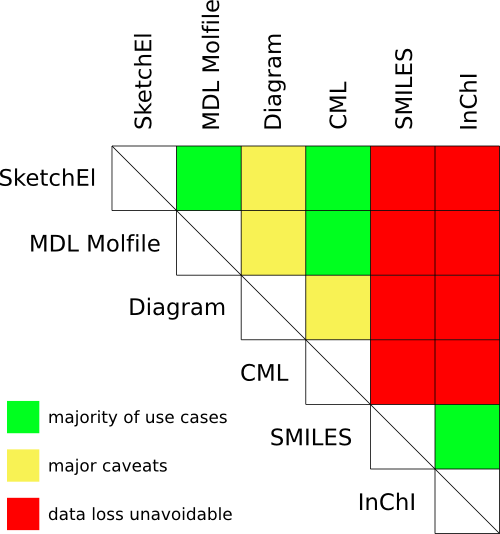
The colour-coding is simple: green means that there is a large and useful overlap between the applicable domains that can be interconverted without any significant issues; yellow means that it is frequently possible with only minor information loss, though very easy to inadvertently stray from the path; and red means that you should never convert between these formats if you want to get your original structure back.
About the formats:
- SketchEl: originally designed for the SketchEl open source sketcher, and the native molecule format used by all products from Molecular Materials Informatics. It is designed to capture 2D diagrams, with a minimalistic and puritanical attention to capturing only well defined concepts, and operating beyond the realm of drug-like organic molecules.
- MDL Molfile: the industry standard for cheminformatics, even though it was originally designed for a handful of bespoke applications with many vestigial features, and major shortcomings, especially outside of the organic subset.
- Diagram: refers to the native file formats of applications designed for drawing chemical structures for the purpose of presentations, meaning that they freely mix chemistry, graphics and arbitrary annotations. Formats include those used by ChemDraw, ChemDoodle, Accelrys Draw, and others.
- CML: the Chemical Markup Language, which is often described as a molecule file format, but in reality it is actually just a loose set of guidelines as to how one might go about encoding a molecule using XML.
- SMILES: a popular line notation format, which omits atomic coordinates, in favour of canonical brevity. Originally designed with the intention of being converted back and forth between a full 2D description, but in practice the domain of applicability is weak, even for the simple organic subset.
- InChI: a more recent line notation format, which provides the broadest domain of chemical applicability for its functional class, but was originally designed only as an endpoint, for disambiguation and database indexing purposes, rather than a way to express structures.
As my introduction would suggest, these file formats are all quite different in their design scope. There are plenty of other atom-based formats that I haven’t mentioned, but in order to avoid getting too verbose, I’m only including those which are designed for use with 2D coordinates or no coordinates. All of them are intended to conveniently capture chemical structures as atoms and bonds, but the primary methods of input (user or algorithm) and deliverables (graphics presentation, algorithmic analysis, database indexing) are quite orthogonal, and quite incompatible with one another.
When converting a chemical structure from one format to another (A → B), the definition of success varies depending on the objective. Sometimes it is quite adequate to ensure that if a chemist were to study A and B, then it would be concluded that they do indeed represent the same species, even though the bytes contained inside the datastructure are different, and the onscreen rendering may be also. Othertimes it is necessary for them to be more literally identical: same atom coordinates, same atom and bond ordering, same wedge bonds, same atom mapping, same style of hypervalent functional group representations, etc. Sometimes it is even acceptable to provide a different tautomer, or even a different protonation state. In other cases it is unacceptable to even lose custom metadata.
What is meant by a fully reliable round trip is more completely described as being able to pick any two formats, starting with a molecule A, and carry out the sequence: A → B → A’ → B’, such that A and A’ are identical, as are B and B’. And what is meant by identical is that when they are serialised and written to files, the files have the same series of bytes.
For the most part the needs of cheminformatics are not quite this stringent: the distinction is whether important data was lost. The definition of important varies, but for any kind of cheminformatics software deployment, it would be hard to argue against the merits of erring on the side of losing as little as possible.
So now lets take a look at how each pairwise permutation works:
- SketchEl ↔ MDL Molfile: for a large use case domain, namely organic structures with common bond types, drawn in an ordinary way without making use of any fancy metadata, these two formats are almost perfectly interconvertible. On the one hand, however, the SketchEl format is designed from the ground up to support a large range of chemistry by the simple expedient of allowing zero-order bonds and explicit hydrogen counting, and it also includes a way to define inline abbreviations within the structure definition, as well as encoding of user defined metadata: all of these capabilities are lost in translation. The MDL Molfile format, on the other hand, has a very long list of features that were designed for specific applications (e.g. query properties, polymer definitions, overall enantiomeric state, molecule name, pharmacophore queries, etc.) and have no exact analog in otherwise related formats. In practice, any attempt to express chemical structures that are not simple organic molecules that follow the Lewis octet rule, or to use non-ordinary features, will result in information loss. The severity varies from trivial to broken.
- Diagram ↔ SketchEl or MDL Molfile: interconversion between any of the diagram formats, and a pure cheminformatics format, is a major problem, because the formats designed for drawing chemical diagrams are typically supersets of the basic primitives required for chemical entities. They provide the minimalistic atom/bond annotation features, but also add a plethora of visual customisation options, graphical objects, and what appears to be higher order markup, such as reaction arrows, block charges, polymer repeat units, etc. Often these additional features are poorly defined for any purpose other than creating pictures, and so they cannot be effectively interpreted by cheminformatics algorithms. Converting a diagram to a cheminformatics format typically works well if the strict subset of atom/bond annotations is adhered to, but use of any other descriptions is quite likely to be uninterpretable. The best case scenario would be for a conversion algorithm to throw an error and refuse to continue, but all too often the conversion proceeds, but with incomplete information (i.e. wrong). Converting into a diagram format is usually a lossless process, though it may lack finesse.
- CML ↔ SketchEl or MDL Molfile or Diagram: converting a cheminformatics format into a Chemical Markup Language document is lossless, since the CML format essentially allows anything and requires nothing. Whether any other software will be able to read it back in correctly cannot be known ahead of time: importing an arbitrary CML document is not reliable, because it may depend on data fields which are not defined as part of any specification.
- SMILES ↔ SketchEl or MDL Molfile or Diagram or CML: the SMILES line notation has been used for a lot of productive cheminformatics work, but its most obvious problem is that it does not include any atom coordinates, so a round trip with any non-line-notation format destroys these. Returning to a cheminformatics or diagram format that has atom coordinates requires the use of a depiction algorithm, and if the original coordinates are recreated identically, this is nothing more than an unlikely coincidence. Many cheminformaticians consider throwing away then recalculating 2D coordinates to be quite a reasonable thing to do, but they are very much mistaken: any chemist who draws out a structure that they went to all the trouble of preparing as part of a their laboratory campaign does not want to see it redrawn in some other style or orientation. Other than coordinates, though, the SMILES format does a lot more damage: functional group standardisation, aromatic ring homogenisation, and general inability to correctly handle all but the most common bonding arrangements mean that this format is only anything other than broken for a narrowly defined subset of organic molecules.
- InChI ↔ SketchEl or MDL Molfile or Diagram or CML: these conversions have the same problems as conversions between SMILES, with some minor adjustments. InChI can operate within a larger domain of chemistry, and has a more general and open method for canonical ordering, so it is a much better choice for using to compare whether two molecules are the same. It was, however, not designed for two way conversions, and performs more aggressive reduction of information (e.g. excluding bond order and disconnecting nonstandard bonds), so even though it has a broader domain, it does even more damage than SMILES.
- SMILES ↔ InChI: this probably works quite well, though there is little demand, since projects usually involve using either or neither of these formats.
The bottom line is basically that there is no such thing as a molecular structure format conversion that does not involve information loss, unless one is very careful to limit the domain of applicability. Any software description claiming otherwise is a misrepresentation. The problems I summarised very briefly above are just the tip of the iceberg: dozens of doctoral theses could be filled with all of the details.
This situation sounds rather bad, and from a certain perspective it is, but there is a fairly simple solution to it: don’t ever throw away the original file! Think of chemical data like a scientist’s original lab notebook. As long as you have that, you can convert anything to anything as much as you want: if you lose something important along the way, just go back to the original, and reinterpret. Remember that, and everything will be fine.

Alex, can you please elaborate on your description of CML? In particular, when you refer to guidelines, do you refer to the overly specific XML Schema, Schematron validation tools, the conventions, or… ?
As far as I can tell, no two programs use CML in quite the same way; if I want to add another bond type, or use different labelling conventions, or dump a whole lot of random stuff in there, it’s still valid. I’ve studied the output from several software packages (since lack of implementation of a standard is pretty much the same as lack of existence), and they are all different enough that I would not describe the results as a format. Just some common patterns of XML usage, but not enough to be a reliable way to transmit molecules between two different programs, unless they make their own private conventions. The mistake CML made is to permit anything: reduces its utility to close to zero. Real value is provided by a format that *disallows* almost everything, but still gets the job done. Much harder to do, but very worthwhile. (And before you ask, I did discuss this with PMR a little while back 🙂 … if the situation has improved, do please post links!)
Reblogged this on ORGANIC CHEMISTRY SELECT.