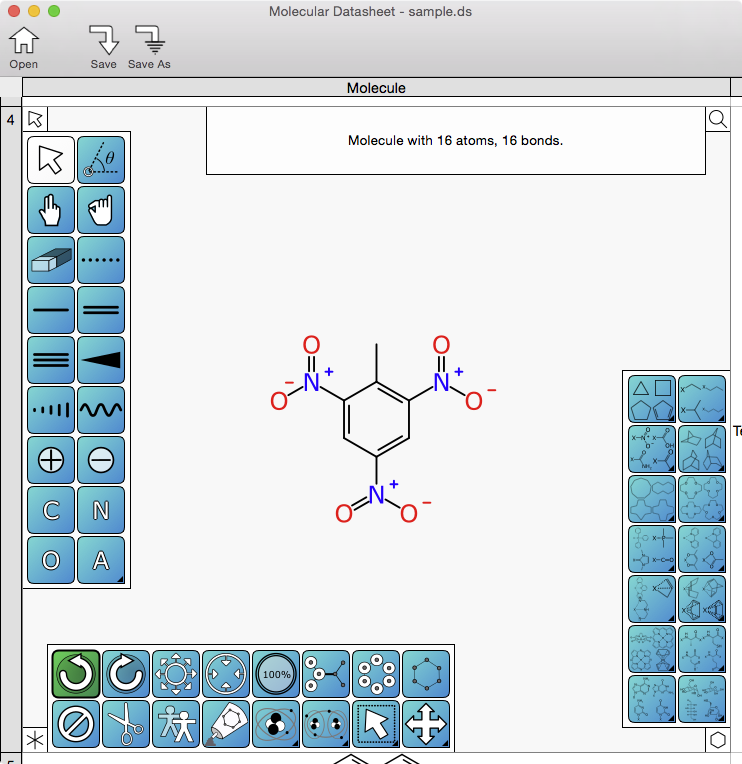
 Since the last sneak preview, the skunkworks project “XMDS” – the Mac OS X desktop version of the Mobile Molecular DataSheet app – has gained enough functionality to make another screenshot, this time showing what the actual molecular drawing interface might look like once it’s done.
Since the last sneak preview, the skunkworks project “XMDS” – the Mac OS X desktop version of the Mobile Molecular DataSheet app – has gained enough functionality to make another screenshot, this time showing what the actual molecular drawing interface might look like once it’s done.
The usual disclaimer applies: there is no timeline for a version 1.0 release, nor that the minimum viable product will look like the early snapshots, but the codebase so far is real enough. From an engineering point of view, the whole thing is built using Swift, Apple’s brand new programming language (for which the current “production” version is not exactly worthy of that designation, but presumably it eventually will be). As is rather implied by the use of a new programming language, all of the code is being recreated starting with a blank slate, though many of the design paradigms draw their inspiration directly from the mobile app codebase of MMDS (which is Objective-C) and the com.mmi libraries (which is Java). As an extra bonus challenge, I have never coded a native Mac OS application before, and Apple’s class libraries for desktop development are not exactly for the faint of heart. While the iOS platform takes quite a bit of getting used to, it has the benefit of designer’s hindsight: Apple had an opportunity to streamline their original ideas when the ported their operating system to the brand new iPhone, and because there are now untold millions of iOS programmers grinding away in Objective-C, the chance of finding a canned solution to any newbie question with a quick internet search is very high. Not quite so much for the desktop platform though, which has a lot more archaic baggage and far fewer end-user programmers.
Now back to the application: the basic operational paradigm is that a window represents a single molecular datasheet, displayed as a table of rows & columns, much like the spreadsheet-like functionality of the open source SketchEl project. For the SketchEl molecular datasheet editor, the spreadsheet and molecule editing are completely different user interfaces. When editing a molecule, a new window appears, like so:
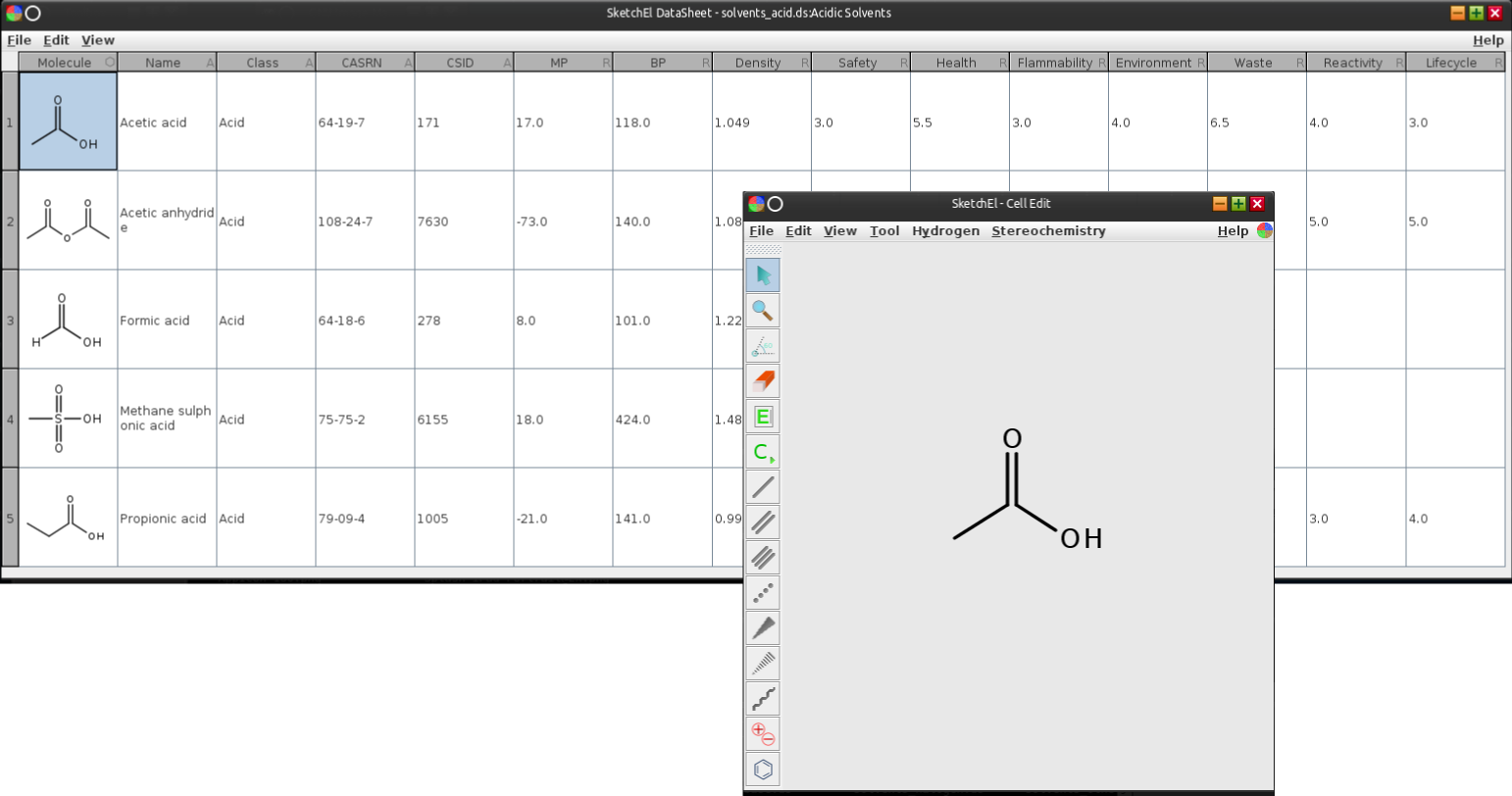
Once the editing is complete, the changes are saved, the window is closed, and context returns to the spreadsheet view. This is a design approach that I’ve never liked, but it is the easiest way to join two modular pieces together. For mobile apps like MMDS, there is also a context switch between editing collections and editing single molecules, but since anything iPad-or-smaller uses the entire screen for every operation, it’s more of a necessary evil than specifically inadequate design.
XMDS, however, takes a different approach. When the molecule editor is activated in the spreadsheet view, rather than popping up a new window, the current cell is zoomed to take up most of the screen:
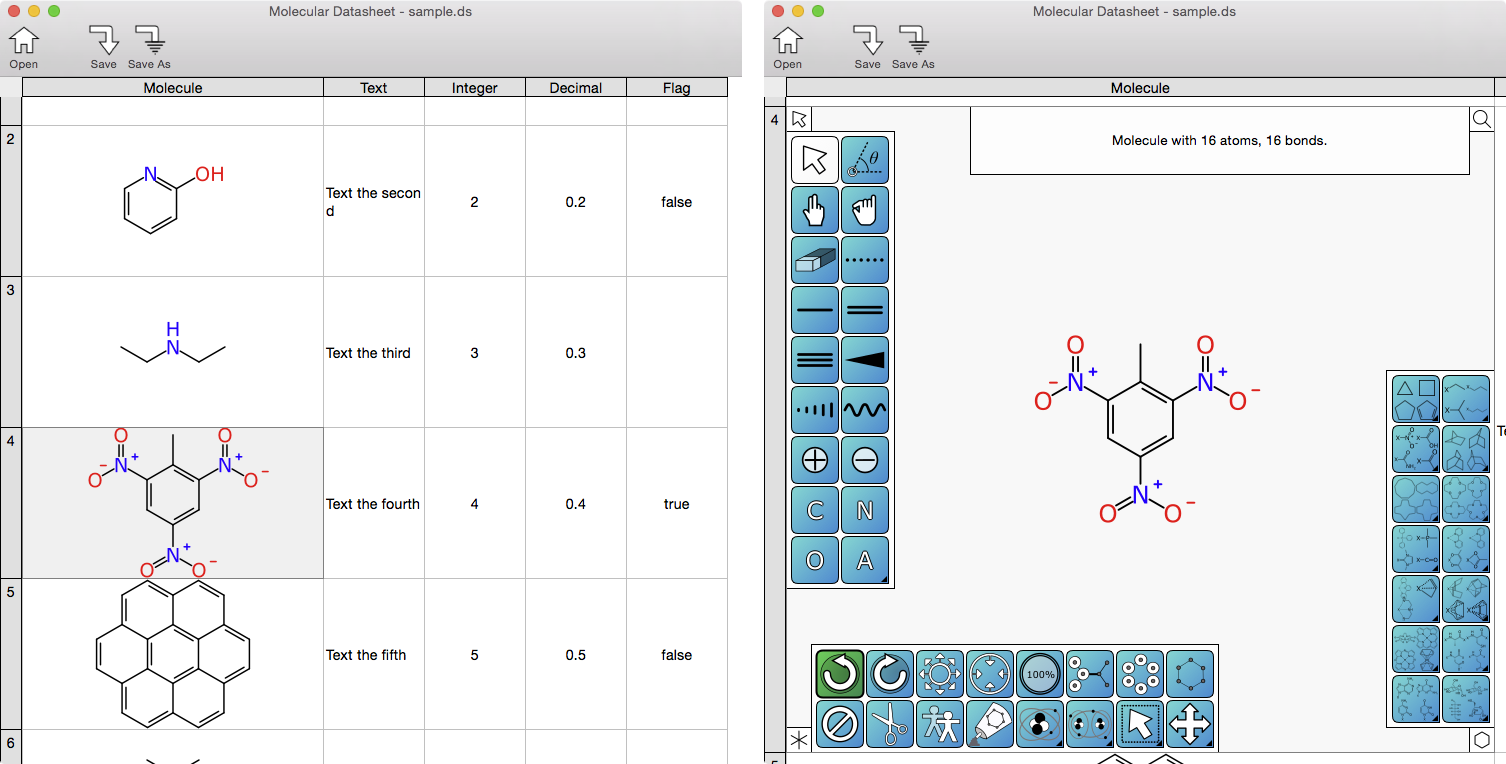
The previously view-only molecule now takes up most of the available space, and presents a variety of panels that expose drawing functionality. Because there is no change of window, the menu bar will remain the same (but with some options enabled and disabled). The editing context is still the spreadsheet: it is still possible to scroll around and look at other stuff while editing a molecule, and features like undo/redo are part of the overall datasheet editing operation, rather than being segregated between two different editors.
Anyone who has used a mobile app from Molecular Materials Informatics will recognise the button style, and some of the icons. The current prototype-in-progress makes 4 different panels available, each of which is tied to one of the corners of the editor cell. They can be pinned, or transiently activated by mouse-over. There will also be a plethora of keyboard shortcuts, and the ultimate goal is to make it quite possible to draw complex structures quickly with only the keyboard – though most users will mainly stick to the traditional mouse-centric approach. In the screenshot above, all 4 of the panels are shown in their default state.
To the left is the tools panel: this is a collection of actions that are familiar to anyone who has ever used any desktop molecular structure drawing interface. The concept goes back to the earliest of drawing tools (predating chemical structure editors), and involves selecting a tool, then heading off to drag the mouse across the canvas in some way, to carry out tasks such as selecting objects, deleting, adding bonds, changing atoms, etc.
At the bottom is the command panel. This is very similar to what is found in the mobile analogs. This style of molecule editing is based on selecting objects then initiating an action, which is carried out entirely by the underlying algorithm. For a clumsy touchscreen this is generally a better way to draw chemicals than the tool style, but for a desktop/laptop computer with a precise mouse-like device, both styles have their pros and cons: which is better depends on the task, and also on personal preference. Sketching is one of those activities where choice is a good thing, because it is so labour intensive and everyone has their own particular needs which vary from session to session. As long as at least one of those choices has a very shallow/nonexistent learning curve, it’s all good.
To the right is the template panel, which is going to work in the same way as it does for mobile, with structure templates being fused to existing structures using a heavy duty placement algorithm. Recently the core mobile technology switched out to showing templates on the right hand side of the screen when used on an iPad, but keeping it as a sub-menu of the main command bank for the iPhone form factor, so even the placement is analogous.
At the top is the detail panel, which will display varying content depending on the selection state. This will also serve as a way to edit individual atom or bond properties in a more manual “dialog box” approach (e.g. a drop-down list for bond order). It will also expose fine grained tweaking of properties, e.g. ways to explicitly specify the X,Y positions of an atom by typing numbers, or changing bond lengths/angles/torsions to a specific value.
As of mid-March 2015, none of this functionality actually does anything, but the amount of infrastructure that needed to be built out in order to make a believable looking screenshot is quite significant, so the remaining pieces may fall into place relatively quickly. Watch this space for more previews, next time a photogenic milestone is reached.

Reblogged this on ORGANIC CHEMISTRY SELECT.