 A two-pack of publications in Journal of Chemical Information and Modeling is now available: Bayesian the first, and Bayesian the second. Both papers are open access, so by all means go read them instead of this blog post. The first paper details the implementation of a variation of the classic naive Bayesian method that is suitable for use with structure-derived fingerprints such as ECFP6 and FCFP6. The text goes into some detail about how it is implemented, to the point of including pseudocode, which complements the fact that the source code is available as part of the Chemical Development Kit (CDK), conveniently and concisely coded up in a single source file. The intention is quite unashamedly to tell you everything you need to know to build the algorithm from scratch, should you be so inclined; and if not, to understand every little detail about how the open source software works. The second paper goes into some more detail about how to use this kind of (“Laplacian-modified”) Bayesian model, including a calibration method, and an extensive study carried out by extracting thousands of model-ready datasets from the ChEMBL database.
A two-pack of publications in Journal of Chemical Information and Modeling is now available: Bayesian the first, and Bayesian the second. Both papers are open access, so by all means go read them instead of this blog post. The first paper details the implementation of a variation of the classic naive Bayesian method that is suitable for use with structure-derived fingerprints such as ECFP6 and FCFP6. The text goes into some detail about how it is implemented, to the point of including pseudocode, which complements the fact that the source code is available as part of the Chemical Development Kit (CDK), conveniently and concisely coded up in a single source file. The intention is quite unashamedly to tell you everything you need to know to build the algorithm from scratch, should you be so inclined; and if not, to understand every little detail about how the open source software works. The second paper goes into some more detail about how to use this kind of (“Laplacian-modified”) Bayesian model, including a calibration method, and an extensive study carried out by extracting thousands of model-ready datasets from the ChEMBL database.
The delivery of the Bayesian model functionality in CDK was done with and on behalf of Collaborative Drug Discovery, and the details of the implementation are equivalent to the CDD Models feature that was delivered a year or so ago. Inclusion of the algorithm in a popular open source toolkit, with a very easy to read and well documented algorithm, and a well defined serialisation format, has been done with the intention of encouraging people to share their models. In the past (or rather, the present) model sharing is rare because model building is generally done with software that is expensive and proprietary, or niche and cantankerous, which means that chances are most readers would not be able to use them. By making a common format that everyone has access to, either as open source or part of a commercial implementation, the excuses for not sharing models will evaporate quickly. Queue in introduction of a new golden age of collaborative activity prediction (!)
As well as the CDK toolkit, and CDD’s Vault product, models can currently be consumed using the Mobile Molecular DataSheet (MMDS) app, by importing the pre-created models. The Approved Drugs and MolPrime+ apps also use the same codebase (which is not the same code as the CDK implementation, but is functionally identical), albeit with the models hardcoded. None of the apps from Molecular Materials Informatics actually allow models to be created on a mobile device, but this is on the roadmap: the algorithm is not hard, rather some thought is required to figure out how to make the workflow sufficiently accessible so that it actually gets used.
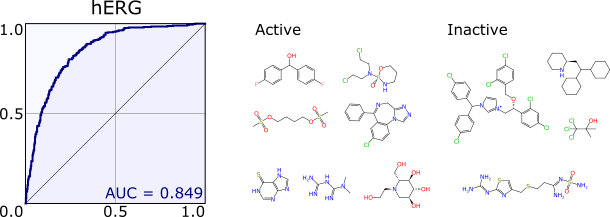
The second paper describes the extraction of a couple thousand datasets from the ChEMBL database (v20), which is a rather awesome resource of well curated structure-activity information. Rearranging the data in a form that is ready to feed into a model is slightly more work than it should be, but the raw data contains all the fields necessary to accomplish this. Having thousands of test cases (with real data) is incredibly handy for validating algorithms, and they were used to put the Bayesian model building through its paces. To do this, it was necessary to design an algorithm for picking a sensible threshold to decide where to draw the line between active and inactive. It’s hardly a trivial problem (read the paper for details).
While this undertaking was explicitly for method validation purposes, the work foreshadows a followup effort to improve the ChEMBL data rearrangement and collation to prepare thousands of models that are useful for prospective drug discovery. Nothing to say publicly about that just yet, but details will be forthcoming soon enough. And this also feeds into a followup project with Collaborative Drug Discovery that involves another derived use of Bayesian models to handle multiple categories, rather than just active/inactive. That will also have to wait until later, but maybe not too long.
From my point of view, Bayesians are everywhere these days: it’s quite remarkable how many corners of cheminformatics support for drug discovery that they turn out to be useful for. No doubt there will be many more applications before this well runs dry.
