 A recent post described preliminary integration of the Molecular Materials Informatics toolkit with KNIME. Further to that objective, some more developments are coming through within the toolkit itself, being used independently of any third party tools: in particular, a way to execute pre-existing workflows with the benefit of a visual interface.
A recent post described preliminary integration of the Molecular Materials Informatics toolkit with KNIME. Further to that objective, some more developments are coming through within the toolkit itself, being used independently of any third party tools: in particular, a way to execute pre-existing workflows with the benefit of a visual interface.
First of all, there is still no user interface for actually editing the workflow files that are used by the toolkit’s pipelining infrastructure. For the moment the only way to design an operation-based workflow for streaming cheminformatics tables from one node to the next is to edit the raw XML file with a text editor, a process that looks something like this:

Obviously this is acceptable only for programmers, and ideally only the subset of them who are intimately familiar with the underlying source code. However, this is not quite as much of a limitation as it may seem, since this workflow system was always intended to be used repeatedly, i.e. design a workflow script to turn A into B, using some collection of steps strung together, and then re-run the script on the same source data or anything else that it applies to. These workflow scripts can be created by an expert, and distributed or customised as necessary.
Scenarios that spring to mind are tasks like washing structures, removing duplicates, calculating properties, sorting, etc. These are very routine tasks in cheminformatics – routine in the sense that they need to be run frequently – but there are so many details that vary depending on circumstances that it is not always straightforward. Being able to easily tailor a specific action sequence involving a series of algorithms, each with its own parameters, is necessary to avoid a lot of manual tedium, and also excellent for repeatability (because if you keep the workflow definition and the original file, you don’t need to remember what you did to it).
A single workflow definition consists of a list of nodes, each one of which invokes a well defined operation, which corresponds to one of the underlying Java classes that conforms to the protocol. These can either be core operations, or anything else that got lumped into the classpath. The nodes are connected together by input/output ports, in a manner that is familiar to any other pipelining software.
There is a special section at the beginning of the XML specification for metadata, and of particular interest are global parameters: these are intended to be provided by the user at runtime, rather than being encoded into the workflow itself. The most common use of these parameters will be things like filenames for input/output, and various configuration options, or in some cases higher order objects (e.g. a molecule to rank similarity to). In a simple contrived example, the file wash.workflow defines two global parameters:
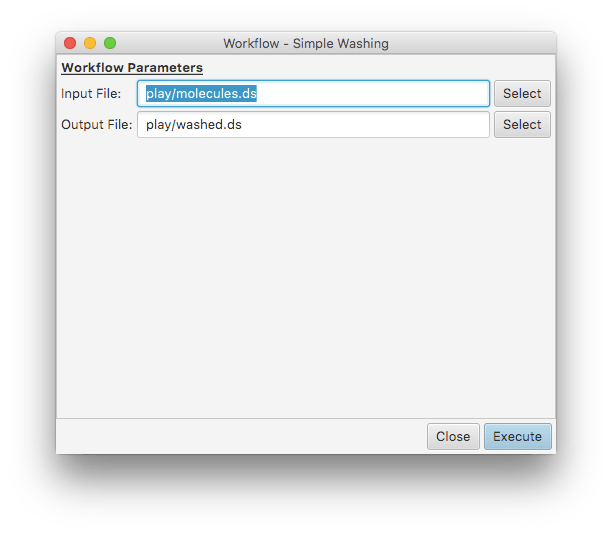
Both Input File and Output File can be specified by the user, in this simple preparation dialog box. They can also be provided using the command line, which either becomes the default option (visual) or the final parameter (when running non-interactively).
Hitting the Execute button brings up a moderately detailed view into what is going on in the workflow:

It may not yet be the most beautiful progress screen, but it informs us that the workflow is made up of 5 nodes, and provides information about how much work each one has done so far. Since this is a true-streaming infrastructure, and most of the operation types are non-blocking, it means that in simple workflows like this one, all of the nodes are running simultaneously. Whenever the supply vs. demand is not perfectly balanced, one or more of the nodes are sitting around waiting to be fed, or waiting for their output buffers to become unblocked.
In the above example, there are two nodes that generate final output content: one of them writes the molecules that have been parsed & washed during the previous steps. When the node is completed, the output file can be launched and viewed on the desktop – in this case, using the XMDS app, which is the default viewer for datasheets:

The honeycomb operation is slightly different, because rather than outputting or writing a molecular datasheet – the universal datatype in this software stack – it creates a vector graphics instance, which is essentially a recipe for converting lines, shapes, text, etc., into some format that can be consumed by presentation software. Clicking on its button brings up a preview:
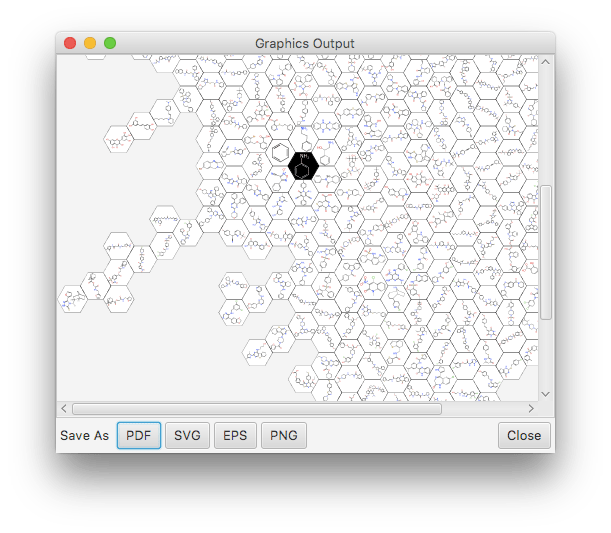
The preview dialog allows the content to be saved in various different formats, 3 of them vector graphics (PDF, SVG, EPS) and one bitmap (PNG).
At the present time, the core Molecular Materials Informatics toolkit is not actually available as a product – you can’t go to a store and buy it – but like all works in progress, there is a roadmap with plenty of milestones, including imminent use by some of my regular collaborators. Because the toolkit is written in Java, it means it can run on any modern desktop/server platform, and most of the functionality is headless, which means it can provide back-end muscle. The feature set is being tailored so that it complements more readily user-facing tools like various apps (MMDS, GLN, etc.) and XMDS on Mac laptop/desktop workstations: there are various integration techniques in the works.
