 The latest unit of progress for the molsync.com site is the addition of an interface for atom mapping. As mentioned in the previous post, basic reaction searching capabilities are operational, but more sophisticated techniques require a way to specify which atoms map to each other on each side of the reaction scheme, in order to describe a transformation.
The latest unit of progress for the molsync.com site is the addition of an interface for atom mapping. As mentioned in the previous post, basic reaction searching capabilities are operational, but more sophisticated techniques require a way to specify which atoms map to each other on each side of the reaction scheme, in order to describe a transformation.
While the R-group method was already operational, properly specific queries require a way to ensure that the atom-to-atom correlations are exactly as you intend them. Atom mapping can sometimes be guessed for reactions where whole molecules are drawn out in full, but when searching, it is common to specify just the interesting parts, and maybe a few other constraints. For example, to find any kind of Diels-Alder reaction, a query with 6 atoms on each side is required:
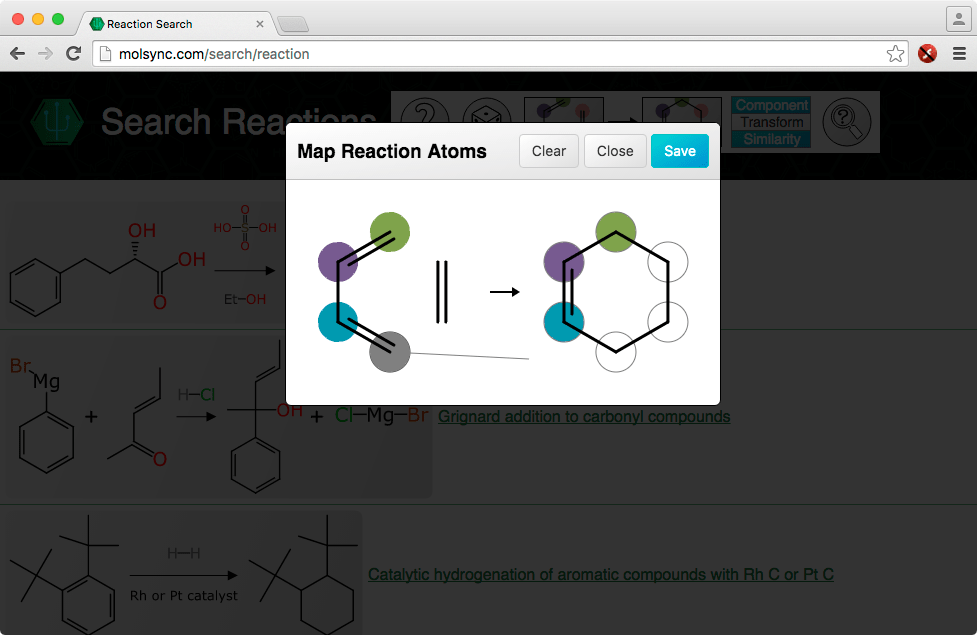
The two sides of the reaction can be drawn with the (still quite new) web-based sketcher, or pasted from the clipboard. What’s new is the ability to click on the reaction arrow between the two components, which brings up the dialog shown above. The atoms can then be dragged onto each other, to indicate the mapping. Rather than showing numbers, the scheme uses colour coding, with a limited palette (and also, when the mouse is hovered over, the connection line is shown).
You don’t need to take my word for it: you can try it out right now, at molsync.com/search/reaction. With the caveat that it’s not mobile-optimised (for that, you need the Green Lab Notebook app).
In this example, using the atom-mapped Diels-Alder transform, and executing a transform search, picks out that results that one might expect:

As you can see, the first screen worth of results shows reactions that are, indeed, of this kind. Just in case it looked like this was too easy, on account of the fact that these results all happen to have the term Diels-Alder in the title, some of the later results don’t have this tipoff:

So far the reaction transform algorithm seems to be holding up quite well, but it is relatively new, and has a fairly sparse validation set, so it will no doubt be fixed up a few times before it becomes genuinely robust. It is quite flexible, in that it can use inputs that have no atom mapping at all (regular substructure), inputs that have R-group (Markush-style) constraints, inputs that have atom mappings, and combinations of the two. The underlying algorithm also supports a number of atom/bond query types, similar to what SMARTS strings are capable of, but these are not yet exposed within the editor, so that counts as a quasi-stealth feature.
There are a couple of followup additions that are high on the priority list. Firstly, the atom mapping interface requires you to manually connect together all of the atoms that make up the transform. It is pleasantly easy to do, but it is entirely stupid. The desktop analog (the XMDS app for Mac OS X) has the ability to “autocomplete” mappings, which saves some effort: it very conservatively looks for ways to add mappings that are pretty much entirely unambiguous, erring on the side of being conservative. This is going to be added to the web UI shortly.
The other odd-one-out feature is the Similarity search type for reactions. Right now it is implemented as just a regular fingerprint ECFP6/Tanimoto search, using either or both sides of the reaction. It does not make use of atom mapping information at the moment – but it will.
Watch this space for further developments, and also new data being fed into the system.
