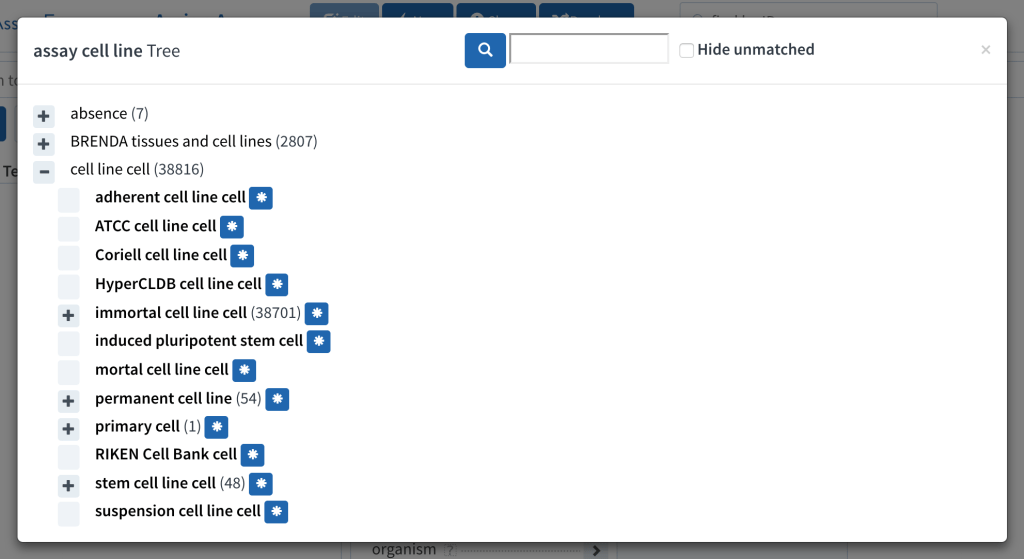
One of the pitfalls of using multiple public ontologies is that sometimes there are two teams doing great work that overlaps, but neither is a superset of the other. This has come up for the BioAssay Express project, which uses both the Cell Line Ontology and BRENDA cells & tissues.
I don’t often post biology-centric articles, but this is an important issue when it comes to using ontologies to describe scientific data. In this particular instance, the Common Assay Template that we use to annotate bioassay protocols has an assignment category called assay cell line. Originally we connected this to a branch that contained imported terms from the Cell Line Ontology (CLO). Shortly thereafter, we brought in the BRENDA terms, which contains terms for a number of cells and tissues.
This seemed like a good start, except that once we began curating content, we very quickly found out that the two ontologies had a large overlap: a number of common cell lines are present in both ontologies. For creating a definitive and canonical way to describe assays machine readably, this is a serious problem: the curator (or machine learning algorithm) is forced to pick either or both of two perfectly valid entries.
It would not have been a problem if one of the ontologies was a superset of the other – we could just throw away the smaller one. But no such luck here: there is a considerable overlap between the two, but they both have a lot of unique entries, as well. And not only that, but they both have their own strengths: the CLO has an intrinsic hierarchy, which is very useful for leveraging the power of ontologies (e.g. CHO clone B cell isn’t just an arbitrary standalone fact, it’s a child of the more general CHO cell, which is in turn an immortal cell line – these things are important to know). The BRENDA ontology that we are using lacks this hierarchy, and is completely flat – but what it does have is a description for each entry, which CLO unfortunately lacks. Therefore even when the terms overlap, we need to transfer vital information from one to the other.
This creates a tricky problem, because we do not have a mapping between the two ontologies, meaning that we have to solve that problem for ourselves. But we can’t do it by editing the ontologies themselves, because both of them are living: we expect to update the source data periodically, and new cells will be added quite often, and deleted or modified slightly less so. If we were to start editing the files ourselves, we would be forking both of these projects: we would lose the ability to conveniently update from the original sources and no longer benefit from the ongoing work of the groups who maintain them.
The problem is tricky but not unsolvable. Connecting up CLO-to-BRENDA can be done with a large degree of automation followed by verification. With the BioAssay Template Editor project (open source), we crafted an algorithm that considers all of the pairwise combinations and calculates a label score (using the venerable Levenshtein metric). Most of the exact matches between cell names, or those with one punctuational permutation (e.g. an extra space or hyphen) are the same thing, and so they can be quickly reviewed by a human with some experience at looking through these things (in this case, me).
Once the algorithm has detected a bunch of candidate pairs, these are proposed in a temporary output file as a patch, which takes a form such as:
# CLO [A549 cell] paired with BRENDA [A-549 cell]
obo:CLO_0001601 obo:IAO_0000115 "Human lung carcinoma established from an explanted lung tumor which was removed from a 58-year-old Caucasian man in 1972; cells were described to induce tumors in athymic mice and to synthesize lecithin." .
obo:BTO_0000018 a bat:eliminated .
obo:CLO_0001601 bat:pairedWith obo:BTO_0000018 .This is RDF/Turtle syntax. The first line is a human readable comment, explaining that the algorithm thought that A549 cell and A-549 cell are probably the same thing. The following line takes the description from the BRENDA term and applies it to the CLO term, and then adds an instruction that tells our template compilation software to disregard the BRENDA term. And the last line adds a note to record that the two terms were paired together, for future reference, so that the next analysis iteration can take that into account.
For this merging process, we basically decided that we prefer to use the CLO URIs because that’s where the hierarchy lives. The BRENDA terms that get matched up divulge their description information. For each match, the total number of selectable items in the BioAssay Express/Common Assay Template is reduced by one, because the BRENDA term is dis-considered, i.e. no longer included in the processed hierarchy.
The actual process of carrying out these matches is not completely clean cut, because it involves back-and-forth between algorithmic matching and human approval. These processes are best done iteratively. In principle there are about ten million pairwise CLO-to-BRENDA matches possible, but obviously they are not equal: those with identical names are generally positive matches, and so they should be proposed first, and thus removed from the pool of candidates for the next iteration. This is why the bat:pairedWith term is added – to make a note to not consider something a second time around. But even that isn’t quite as simple as it seems, because the CLO does have some duplicates with the same name, hopefully in different hierarchies: and so at the discretion of the available human, there can be one-to-many or many-to-one matches.
Once the scoring is relaxed to allow not-quite-identical labels, the degrees of freedom for results blows up. It becomes useful to curate the proposed matching options and reject some of them, which can be done by switching the parity of the mapping instruction to bat:notPairedWith. This means that for the next iteration, that match will not be considered as an option, and instead both of those terms may be paired with something else instead, possibly a match with a less-similar name.
This process of coming up with a semi-automated matching workflow, then taking the best characteristics of both ontologies, is a bit involved, but it has highly desirable properties. The main one is robustness: we can update either of the ontologies anytime, and the deliverable – a file called cellmerge.ttl – will be applied as a patch when the ontologies are recompiled.
This is only possible because we made all the modifications we need for our own purposes without changing a single line from the source ontologies.
Furthermore, we can re-run the algorithm as many times as we want, looking for new matches. And we can easily delete old ones if they turned out to be wrong, and the original content will magically reappear. In the first pass, we found almost 500 correspondences between CLO and BRENDA, and anyone can download the file and leverage these. Just download the cellmerge.ttl file and look for bat:pairedWith if you want to use our work to build a mapping between the two. And by all means report any mistakes or additions.
The work is also ongoing. We will consider new ways of finding pairs between the two (e.g. we haven’t tried processing alternate synonyms yet, which may yield another batch). Any further results we obtain will also be available on the open source repository as soon as we get them ready for use within our own projects.
On a closing note, it would be nice if ontology creators would organise collaboratively around topics, but the reality is that projects with different priorities overlap for all kinds of reasons, many of which are unavoidable. Quite often the consumers of these ontologies are stuck with the fact that there’s no way to get all of the value from just one. The process I’ve described for cell lines is a pragmatic compromise to achieve this with modest effort, and it can be done without breaking anything.
