 An important milestone in has been reached in the migration of complicated structure-based calculations to pure mobile. The latest version of MMDS (1.5.9) is now available on the AppStore, and allows visualisation of calculated properties for individual molecules, as well as calculating new columns for entire datasheets.
An important milestone in has been reached in the migration of complicated structure-based calculations to pure mobile. The latest version of MMDS (1.5.9) is now available on the AppStore, and allows visualisation of calculated properties for individual molecules, as well as calculating new columns for entire datasheets.
The previous post described how recent porting of core technology (e.g. substructure query fragment searching) to Objective-C and iOS has opened the door to a variety of calculation types, including atom type-based contribution methods, while the post before that described how the porting of modern fingerprint types has enabled Bayesian models to be used. These progressions are significant, because the previous method of choice for carrying out difficult (or resource intensive) calculations was to hand off the data to a webservice, and await a response. The two technical arguments in favour of taking this approach are slowly but surely eroding: as device capabilities improve, the performance argument becomes less compelling, and as the difficult algorithms are migrated to Apple’s unique and incompatible-with-everything-else development tools, that stops being a problem as well.
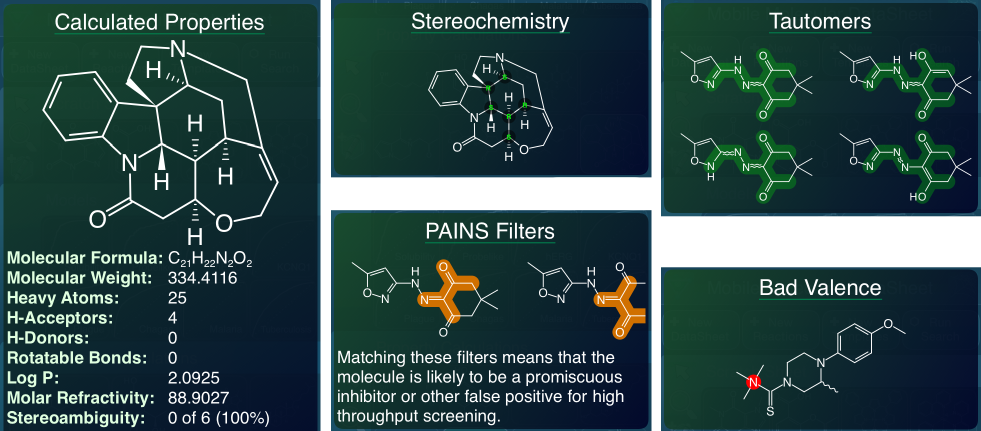
The current version of the Mobile Molecular DataSheet has had two major features retrofitted: bringing up the calculation panel for an individual molecule now displays a wealth of calculated information, some of it in the form of numbers, some as graphically annotated structures. The calculation panel for whole datasheets offers the option for calculating scalar properties for each row, and storing the results in columns, which means that MMDS can be used as a calculation engine for other uses (e.g. QSAR or various kinds of visualisation). The individual molecule property calculation uses the same code as the MolPrime+ app, which received these new features first (but the second instalment is still awaiting review and should be available soon).
The properties that are now available and can be calculated locally on the app, with no internet connection or security conerns, include:
- Easy-to-calculate scalar properties: molecular formula/weight, # heavy atoms, H-acceptors/donors, # rotatable bonds.
- Log P & molar refractivity: both calculated by an atom contribution method (published by Crippen way back when), which requires implementation of substructure searching.
- Bad valences: reviews the valence counts for each of the main group atoms and reports egregious mistakes (e.g. pentavalent carbon).
- Stereochemistry: sites for R/S and E/Z stereochemistry are identified and their labels calculated, with unspecified or known ambiguous cases classified appropriately.
- Tautomers: common H-shifts are identified and the complete list of tautomeric forms are enumerated, with duplicate equivalent molecules removed, and racemised stereocentres labelled accordingly.
- PAINS filters: the original set of queries for identifying frequent hitters and other high throughput screening problem compounds is applied, and any matches are identified.
- Mass distribution: the isotope distribution is calculated for integral masses, as well as the exact mass for the base peak.
The presentation of these calculated properties varies for single molecule vs. whole datasheet modes. Some numeric properties are displayed as such, alongside the structure:


Properties that are not inherently scalar are shown as structure overlays, for example valence mistakes which are identified in red:

Stereochemistry is also shown with the labels overlaid on top of the structure diagram:
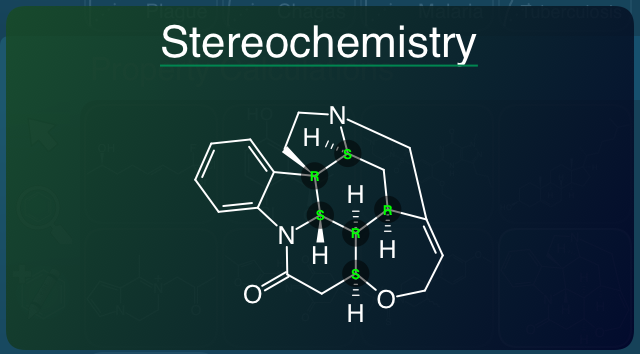
However, it also induces a descriptive field, for counting the number of known/unknown stereocentres, and also the concept of “stereoambiguity”, which is essentially 2-N, where N is the number of unresolved stereocentres (so a compound with one unlabelled chiral or alkene-like stereocentre would have a value of 0.5, whereas a compound with two would be 0.25, etc.). This is the beginnings of an idea referred to as “confidence in chemistry”, which you are likely to be hearing more about soon from Chris Lipinski.
Tautomers are presented as an enumerated collection of compounds, when they apply:

Visually they are shown as a scrollable graphic, and in each case the atoms that are affected by a tautomer shift are highlighted. Any stereocentres that were distinctive in one of the tautomeric forms but have been the subject of one of the tautomer shifts are denoted as being ambiguous. For numeric purposes, a similar idea of “tautomer ambiguity” is calculated, with then confidence being 1/N, where N is the total number of tautomeric forms.
It should be noted at this point that some of these properties take some time to calculate. When the property viewing dialog is opened, it initiates a background thread, which grinds away at producing the results. Some of the properties are fast (e.g. molecular formula), some of them are just slow enough that they would glitch the user interface if they were not put in the background (e.g. log P requires a number of substructure matches), and some of them can take seconds, depending on molecule size and how new your device is, which in particular applies to the PAINS filters. These are built from a collection of SMARTS strings (upconverted to connection table queries, with the meta-sub-fragments expanded out, the total is close to a thousand) which all have to be run against the current molecule. The dialog panel updates as and when each of these becomes available:

As for tautomers, the PAINS matches are rendered for individual molecules as a side-scrolling collection. Most compounds hit zero-or-one of these filters, but it is possible to create molecules that hit many of them, and symmetry can crank it up further.
Calculating properties for a datasheet involves a less graphical setup dialog:
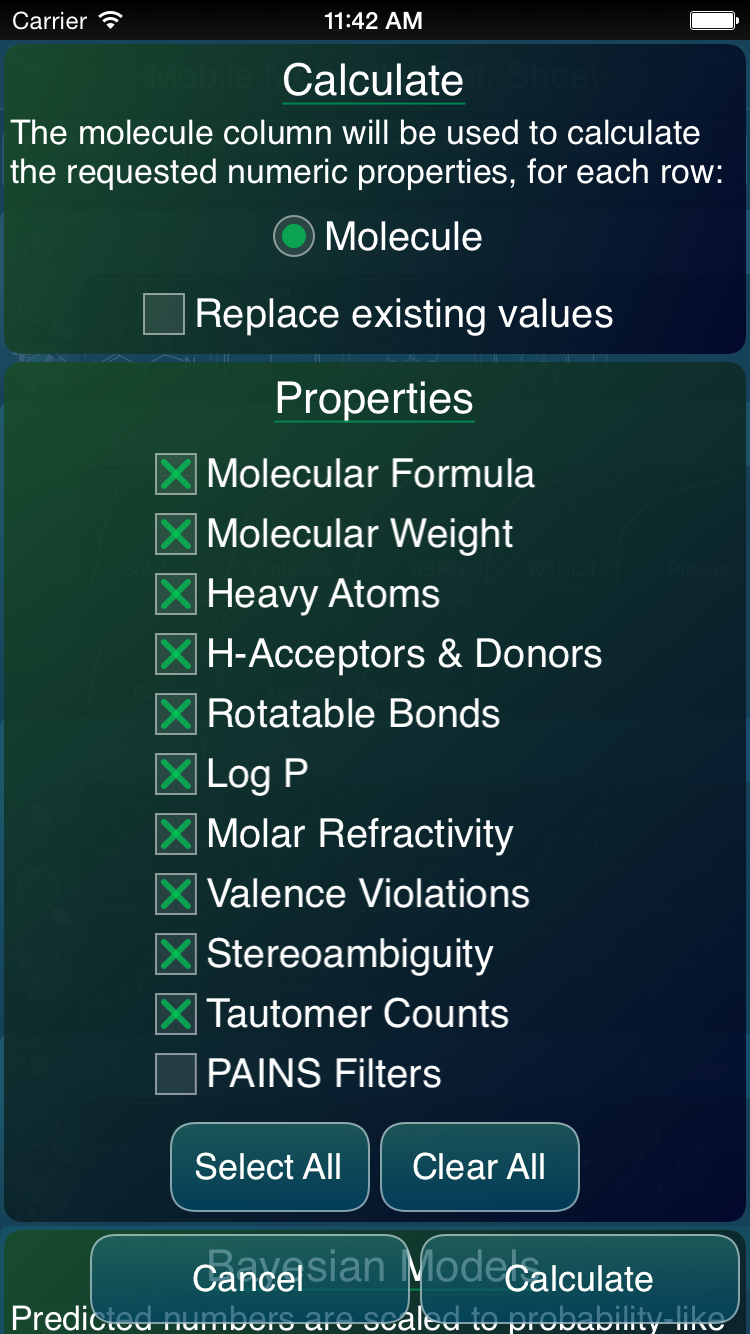
It consists of a checkbox for each of the properties that are desired. By default everything is on, except for the slowest calculation type (PAINS). Note also that the screenshot above shows an obscured section underneath, with the heading: Bayesian Models. This is the next major extension to the MMDS app, and it’s coming soon.

And a more official documentation page to go with it: http://molmatinf.com/propcalc.html
Just FYI
http://www.macinchem.org/blog/files/2269bda317c944fe18e04996a885eb39-1665.php
Cheers
Chris >
Reblogged this on ORGANIC CHEMISTRY SELECT.