 My latest publication has just come out as early access in Journal of Chemical Information & Modeling, entitled “Open Source Bayesian Models: 3. Composite Models for Prediction of Binned Responses“. This is an extension of previous work on the Bayesian/fingerprint theme, and in the interim while waiting for peer review, we have some additional developments to share.
My latest publication has just come out as early access in Journal of Chemical Information & Modeling, entitled “Open Source Bayesian Models: 3. Composite Models for Prediction of Binned Responses“. This is an extension of previous work on the Bayesian/fingerprint theme, and in the interim while waiting for peer review, we have some additional developments to share.
This most recent paper is co-authored with my regular collaborator Sean Ekins and Krishna Dole of Collaborative Drug Discovery (CDD). The backstory is that I’ve been frequently working with CDD for some years now, and we have had reason to implement fingerprints (here) and Bayesian modelling (here & here) functionality and make it available as part of the open source Chemical Development Kit (CDK).
In various projects, experimental or otherwise, we have gotten a lot of mileage out of the basic Bayesian modelling method, and one of the things that we like about it very much is that it is a relatively hands-off method. This means that models can be built and applied with minimal operator expertise, which makes everything much more agreeable when one’s mission is to help busy scientists without adding to their burdens. They are also benefit from being intuitively interpretable, given that we choose to feed in fingerprints that are derived directly from the structural composition (ECFP6 in particular).
The key limitation of the basic naïve Bayesian method is that it is inherently two-state. The content that is fed into the model has to be precategorised as active or inactive, which is good enough for many projects, but far from all. The Bayesian method does produce a continuous value, but it cannot be directly adapted to modelling continuous properties (or more accurately, it can but should not: the results are demonstrably awful). If the target is more along the lines of high, medium and low, or more than that many categories, then Bayesian does not apply.
An idea that we began exploring awhile back is to divide a set of continuous activity datapoints into bins, e.g. a series of pKi values from 9 to 1 might be divided into 4 bins that cover the ranges [9..7, 7..5, 5..3, 3..1]. Once the values are all reclassified as being discrete, i.e. their assigned bin, there are plenty of ways one might imagine that a model could try to estimate which bin some arbitrary molecule belongs to. If you can draw a molecule, and it can guess the right bin, then you walk away knowing the ballpark of activity.
In our early experiments, it turned out to be quite effective to build one Bayesian model for each bin (i.e. active = in the bin, inactive = not in the bin), and then using these models to competitively predict which bin scores highest. The biggest problem turned out to be that the method is extremely sensitive to choice of binning boundaries. This makes sense when you think about it, because if you draw the boundary lines through the middle of regions where molecules have similar structures and similar activities, then predictivity will be very low, whereas if distinct clusters of similar activity/similar structure are allocated to different bins, then predictivity will be very high. And so for this reason predicting nice round numbers for bin boundaries (like [9, 7, 5, 3, 1]) turns out to be a bad approach because there’s no way of knowing if SAR clusters are going to be separated well or badly.
This project got picked up again after we invented some new tricks, which was motivated by our large scale processing of the ChEMBL dataset (see reference): we had thousands of datasets, and we needed to figure out a way to pick a threshold between active:inactive, without having to study the data manually. We came up with a rather effective method that evaluates potential thresholds by playing off the population gradient, size balance and the metrics of a trial Bayesian model.
It works very well for feeding into two-state Bayesians, and so we did a bit more work to see if we could adapt the method for multi-state, i.e. detecting boundaries for multiple bins, such that the following step (competitive Bayesian models) would be as effective as possible. That’s the summary in a nutshell: you can read the paper for all the details.
We made the underlying algorithm available to the world as open source, via GitHub, before we submitted the paper. Peer review in old fashioned journals being what it is, quite a lot of Internet time went by while it was being evaluated. In the meanwhile we were not idle: we have since put together a basic user interface for the tool, and now made that available, too, also via the GPL v2 license. It is written in Java 8/JavaFX, and so will run on any of the 3 workstation platforms (Win/Lin/Mac). The bottom line is that you can now use it without writing any code:

The above snapshot shows the UI once it has been fed with a single file containing activity data, and instructions to reserve 10% of the records to make a test set.
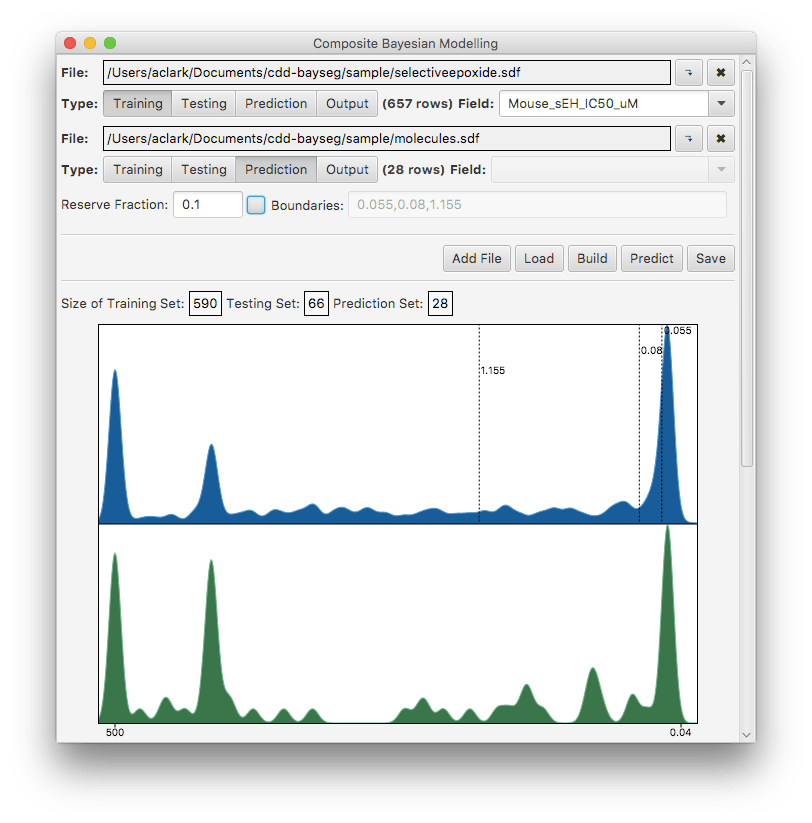
After hitting the Build button, the first change is that the bin boundaries are annotated: in this case, the algorithm decided to divide the content into 4 bins, as marked above. These may not look entirely intuitive in terms of splitting clusters of activity – because what it is doing internally is playing this off against differences in structural composition (which of course you can’t see on a simple population histogram).
Below the population graphs are the cross validation matrices:

These are described in more detail in the paper, but the salient point is that more values along the diagonal is better: a diagonal hit means that the highest scoring bin was the correct one. Off-diagonal hits means that the highest scoring bin was off by one or more. This is summarised underneath by showing the enrichment, which is the hit rate relative to tossing a coin (assuming all options are equally likely).
The real action starts when feeding in molecules upon which to make predictions:
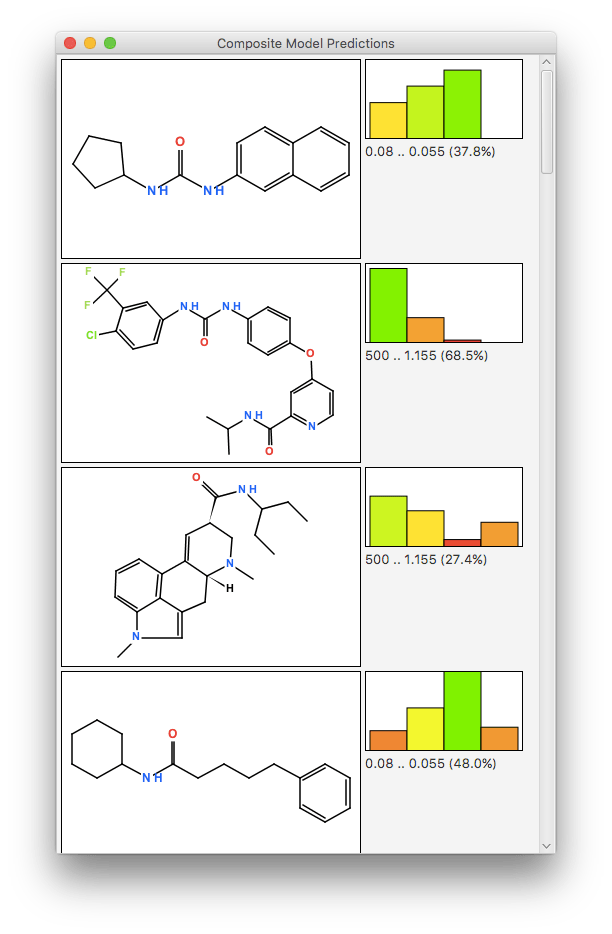
Visualising the results fully requires more than just a single scalar: ideally a prediction should be evaluated by looking at the prediction scores for each of the bins. When one bar is at maximum height, and the other bars are all very low, this corresponds to a high confidence prediction. There are plenty of cases where a molecule has structural features that makes it look like it could easily belong to more than one bin, or doesn’t really belong to any of them.
Besides visualisation, the interactive tool allows prediction results to be saved to an output SDfile, which includes all the relevant information about the bins and the predictions, so it can be analysed by other software. The tool is also designed to work on the command line as well as interactively, so it can be incorporated into scripts.
You can check out this tool anytime – it’s available to the world on GitHub. You can either compile it yourself, or just grab the latest .jar file and run it as-is. And keep in mind that we will continue to work on the project, by way of fixing bugs or occasionally adding a feature. If it’s helpful, or almost-helpful and you’d like it to be better, then do please get in touch and let us know.
The purpose of this interactive application is in large part to serve as a prototype for implementing a new piece of functionality within the CDD Vault ecosystem. Downloading and using the open source application is all very well, but you need to organise your own data going in & coming out. Once the equivalent functionality is available within the CDD web-based platform, where your data already resides, we can make the user experience all just work without any extra hassle.
