 The CDD Vault web product has recently delivered on one of its promises: making models truly sharable to the community, including both open source and participating commercial products. Bayesian models can now be exported in a well documented and openly consumable format, which removes a major bottleneck in the creation and use of models for structure-activity predictions.
The CDD Vault web product has recently delivered on one of its promises: making models truly sharable to the community, including both open source and participating commercial products. Bayesian models can now be exported in a well documented and openly consumable format, which removes a major bottleneck in the creation and use of models for structure-activity predictions.
As is often the case in this field, the story begins with a technology vision: to take the successful and popular algorithms for structure-derived fingerprints (ECFP6 and FCFP6) and Bayesian modelling, which were until recently much more difficult to use without spending a lot of money or writing a lot of code, and make sure that anyone who wants to use them can do so with open source tools or with commercial implementations of the same algorithms and file formats. The emphasis here is on “same“: by making models and the algorithms that are required to use them identical across many platforms, toolkits and products, this removes the main technological roadblock for model sharing. One of the major excuses for not sharing models upon publication is that only a small subset of readers would be able to use them, which has never sat well with many of us. But there’s only so much complaining one can do when there isn’t a viable alternative, which is where Collaborative Drug Discovery (CDD) came in, by securing a research grant and commissioning the building of the tools necessary to provide one.
Implementing a variant of the ECFP6 and FCFP6 fingerprints (which cannot be reproduced from the original algorithm because it withholds trade secrets) was done with particular care to ensure that they are very easily portable between platforms. The reference implementation was submitted to and is now part of the Chemical Development Kit (CDK), and was quickly ported to Objective-C/iOS in order to be used by the TB Mobile app. Around the same time, CDD Vault was augmented to allow Bayesian model building, which uses the open source fingerprint implementation with its own Laplacian-modified naive Bayesian algorithm, and a custom-built HTML5 interface. This represented a major step along the road to bringing this flavour of model building to a larger audience, but the next step had to wait for a few more pieces to be put into place. Recently, we released a full implementation of the abovementioned Bayesian algorithm, and a well defined file format, for inclusion in the CDK library, to follow up on our fingerprint contribution. There is a paper in the Journal of Chemical Information and Modeling which describes this in great detail, so no need to repeat the specifics, but the important part is that the codebase has been used to further extend CDD Models so that it can produce an exported deliverable. Which in this case happens to be a downloadable file with the suffix .bayesian, which can be used by several algorithms that exist in the wild.
This is important to Molecular Materials Informatics for one very important reason: my company’s product lineup has a variety of ways to put Bayesian models to use with workflow scenarios that make sense, namely for mobile apps, which tend to have a more casual and transient use pattern than most other platforms. The Mobile Molecular DataSheet (MMDS) app can import and use Bayesian models in several interesting ways, but until a few days ago, there was no really convenient way to build those models, unless you were willing to work with toolkits and do a little bit of programming. But now there is, and it works rather well.
To illustrate, I will show an example that uses three products, from desktop to web to mobile:

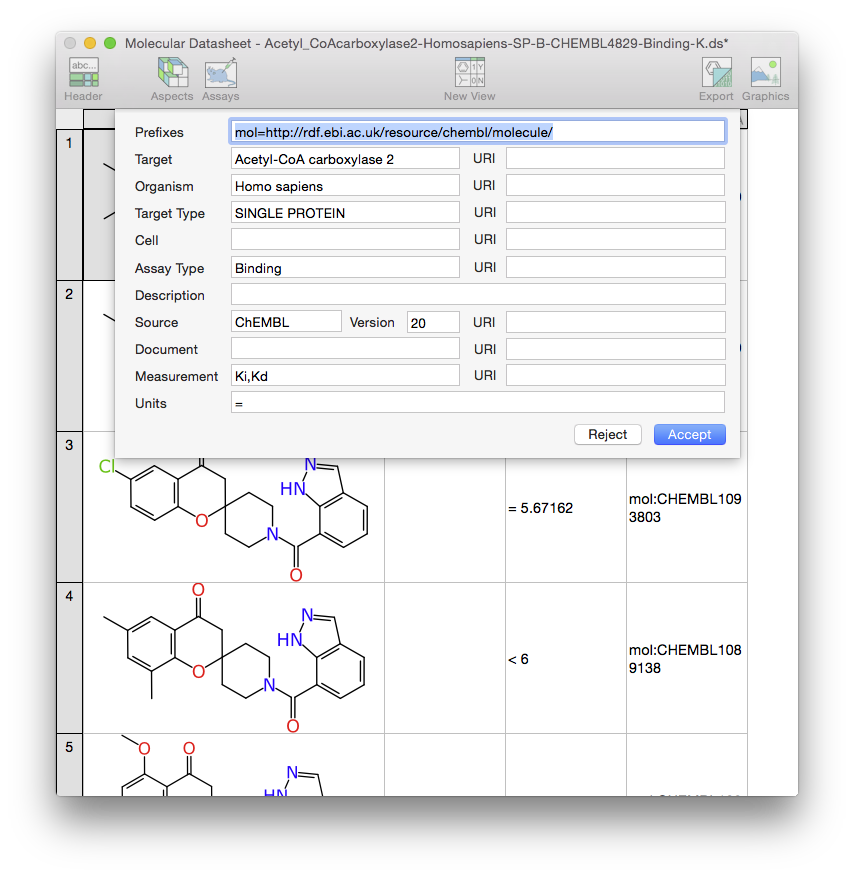
The above snapshots show the OS X Molecular DataSheet (XMDS) desktop app in action (which is in beta if you are interested in trying it out). The datasheet being added is an extract from the ChEMBL collection, which includes all assays of a certain type against a certain target (built by a method described in Part 2 of our recent Bayesian articles). The second screenshot highlights the fact that the datasheet is marked up with the AssayProvenance aspect. While it happens that this particular example data collection was created by a script from preexisting data, the XMDS app is designed with the primary purpose of being a content creation tool, so were I the scientist responsible for producing this data, it would’ve been the ideal place to manifest its digital representation.
What the XMDS app lacks right now is the ability to create Bayesian models, but what it does have is a convenient way to get a collection of data uploaded into CDD Vault:
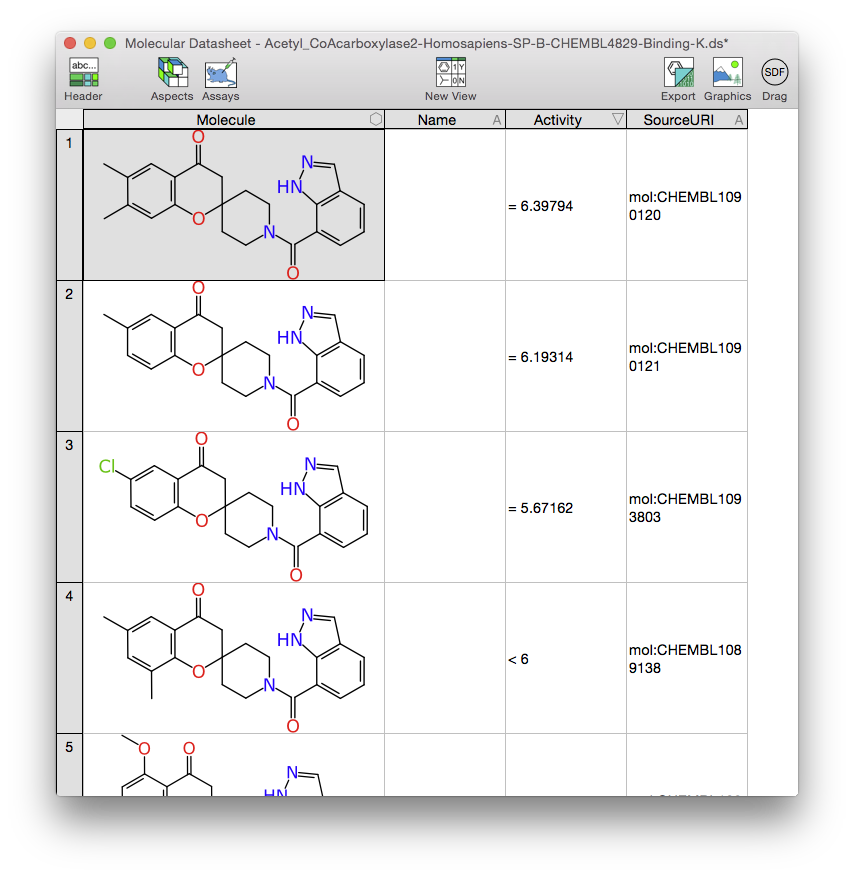

The two images above summarise a couple of steps, but the long story short is that the data transfer is a drag operation from the XMDS app into the upload button of the browser web page that is running CDD Vault. It may seem like a small convenience, but drag-n-drop can be a lot less painful than exporting an SDfile to some file folder, switching to the browser, hunting around for the file to upload, finishing the operation, then remembering to delete the file later rather than leaving superfluous junk lying around.
After that it’s a matter of going through Vault’s import process for bringing in the SDfile that XMDS created for purposes of the transfer:
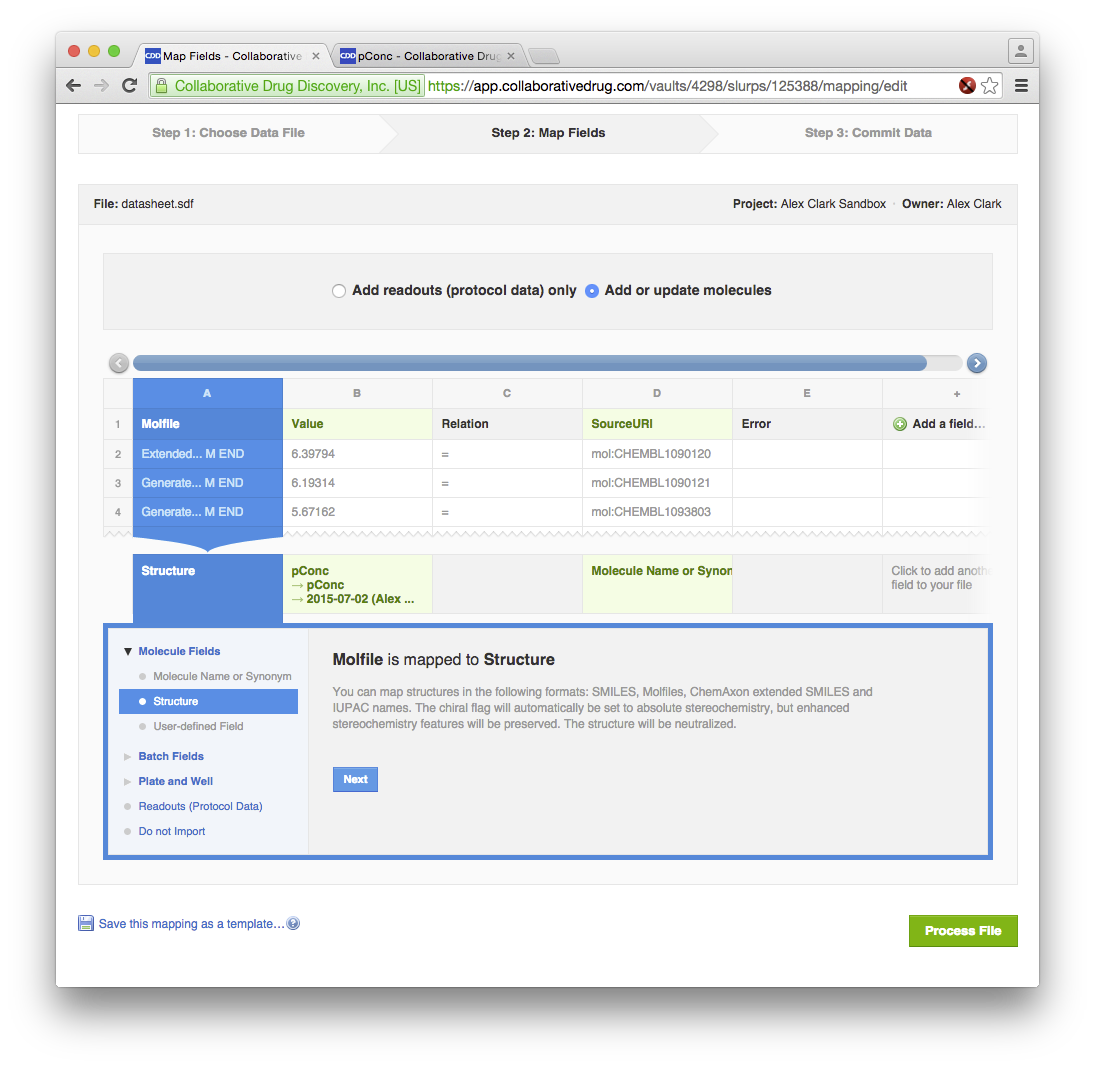
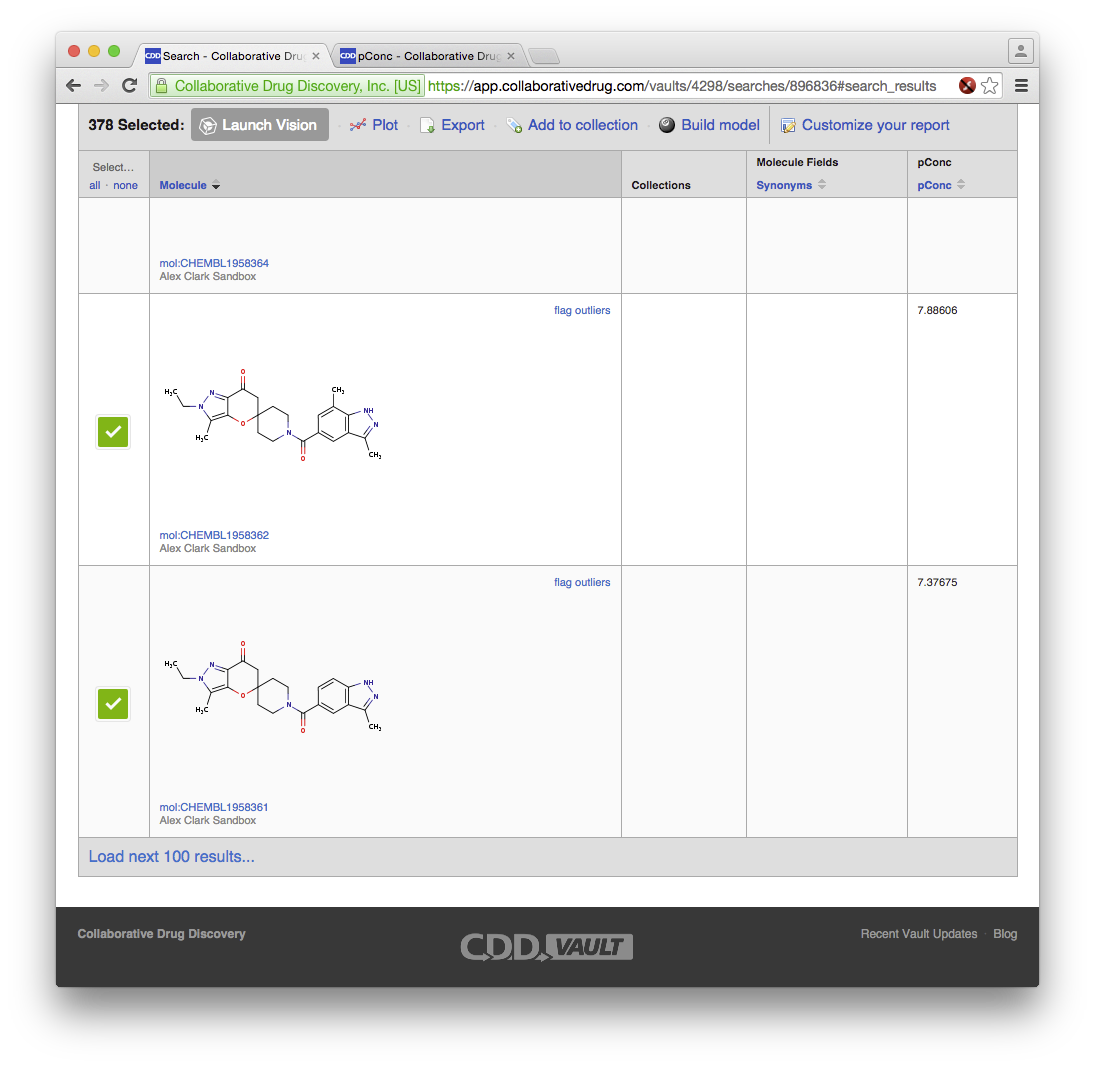
At this point, the data is now stored rather securely on the cloud. Besides looking at the molecules, there are several other visualisation options, such as taking a quick look at the activity distributions:

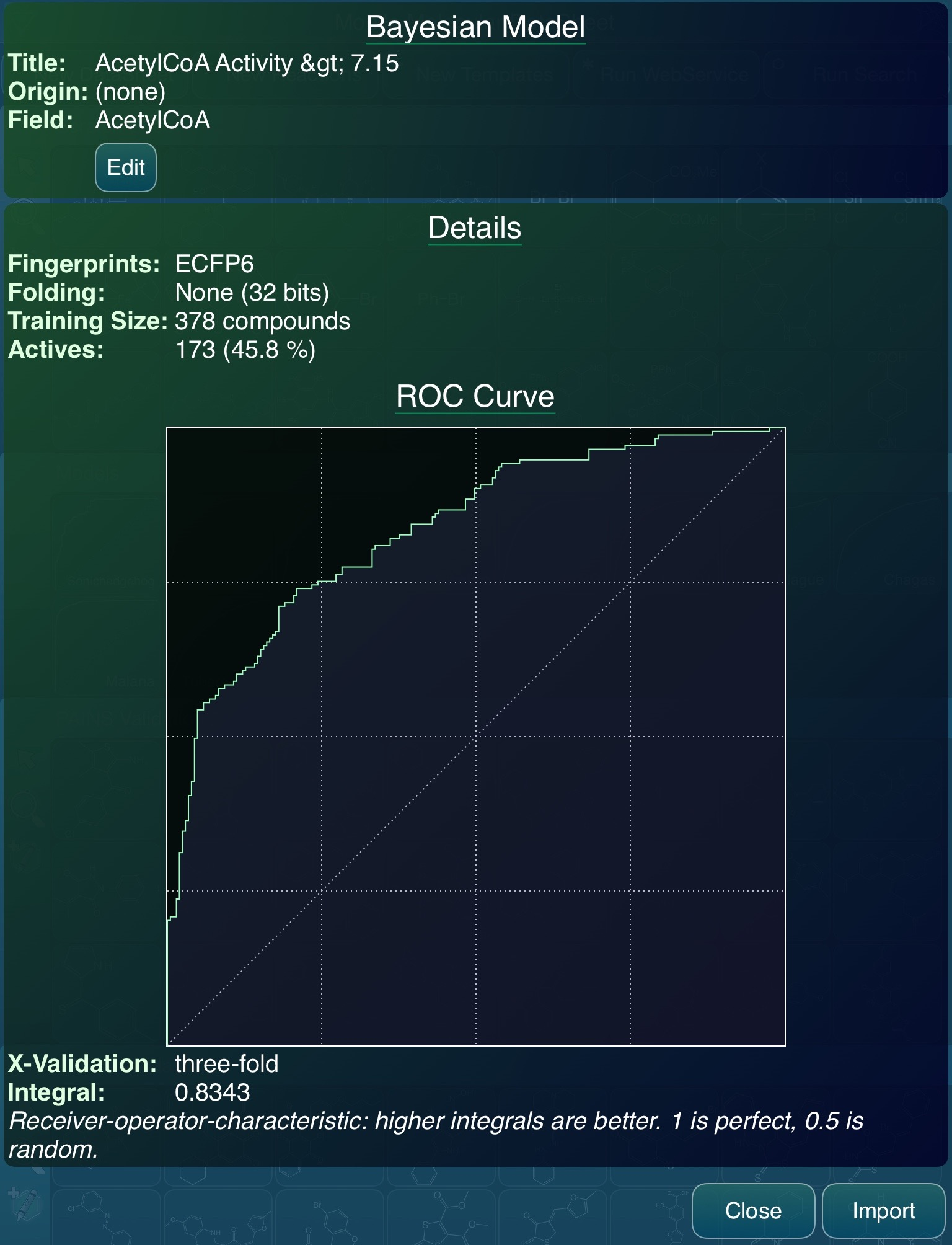
The above plot looks about right, since the activity values are in pK scale (i.e. -log10 of concentration), where a value of 6 equates to 1 micromolar. To build a Bayesian model, it is necessary to convert this into a true/false partition by defining a threshold, which in Vault is accomplished by using the search interface. Deciding on a threshold can be a bit of a black art, and in this example I’ve “cheated”, because part of the process by which the data was extracted from ChEMBL involved using a state of the art algorithm for determining the best cutoff threshold for Bayesian model building purposes (also described in the Part 2 Bayesian paper), and for this reason I already knew that 7.15 was a good threshold:

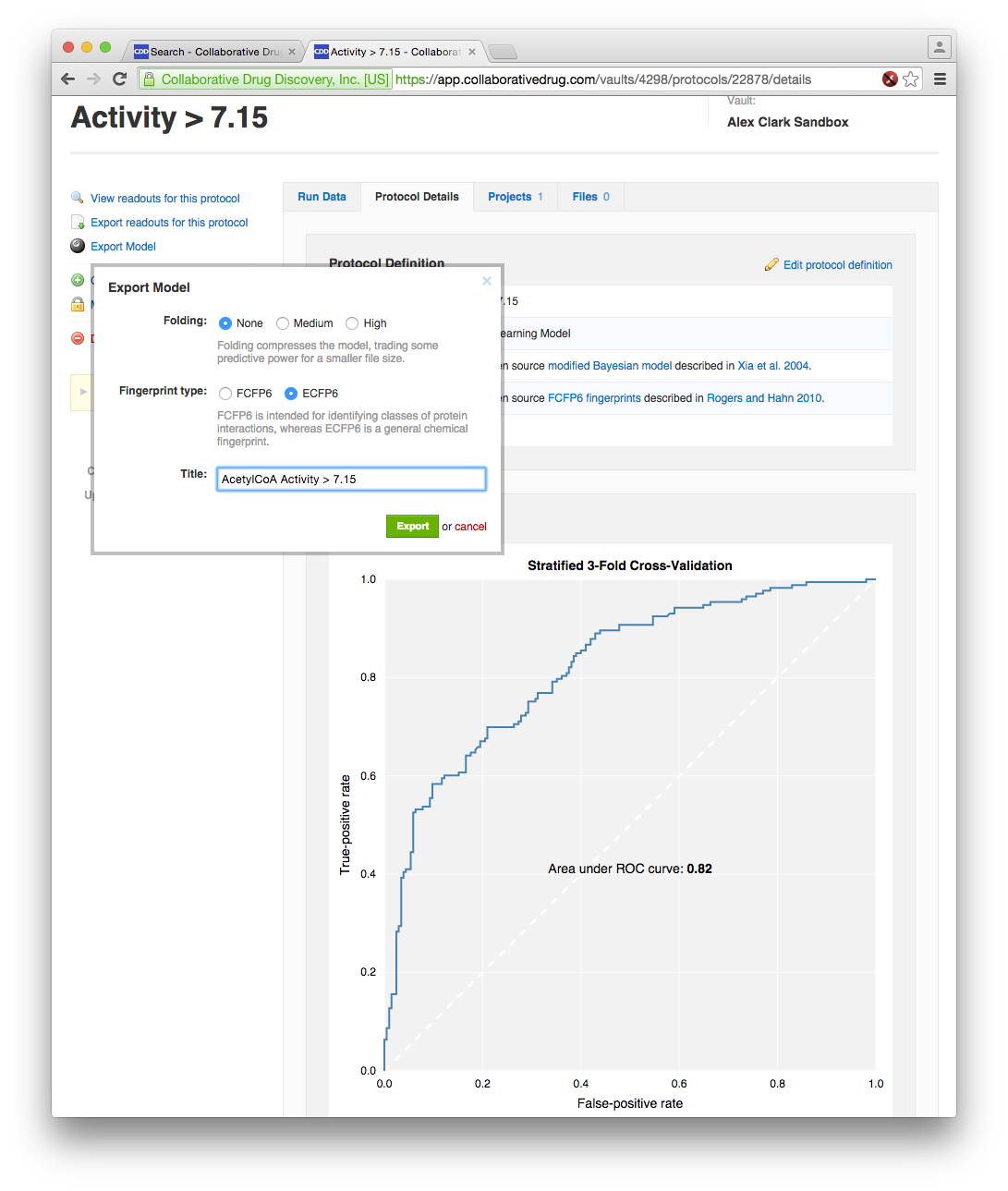
The above rendition of the receiver-operator-characteristic (ROC) curve is the CDD Models implementation of Bayesians. From the point of view of this article, the more interesting of these is on the right, as it shows the export feature. This is where the model is converted from the CDD-specific representation (that applies only to Vault users) to a serialised representation that is subsequently downloaded as a file, that can be used by any compatible software, anytime, anywhere. There are a couple of questions to answer, since there are several sub-flavours (e.g. ECFP6 vs FCFP6; fingerprint folding – compact vs. detailed), but it could hardly be much simpler.
Once the model file is available, it can be consumed. If Vault is being used on a mobile device (iPhone or iPad) it can be opened directly with the Mobile Molecular DataSheet (MMDS) app; or if, like with this demo, it was being used on a regular desktop computer, the file can be transferred any number of ways. My personal favourite is copying it to a Dropbox folder and launching it into MMDS directly from the Dropbox app. Either way this triggers the import dialog:
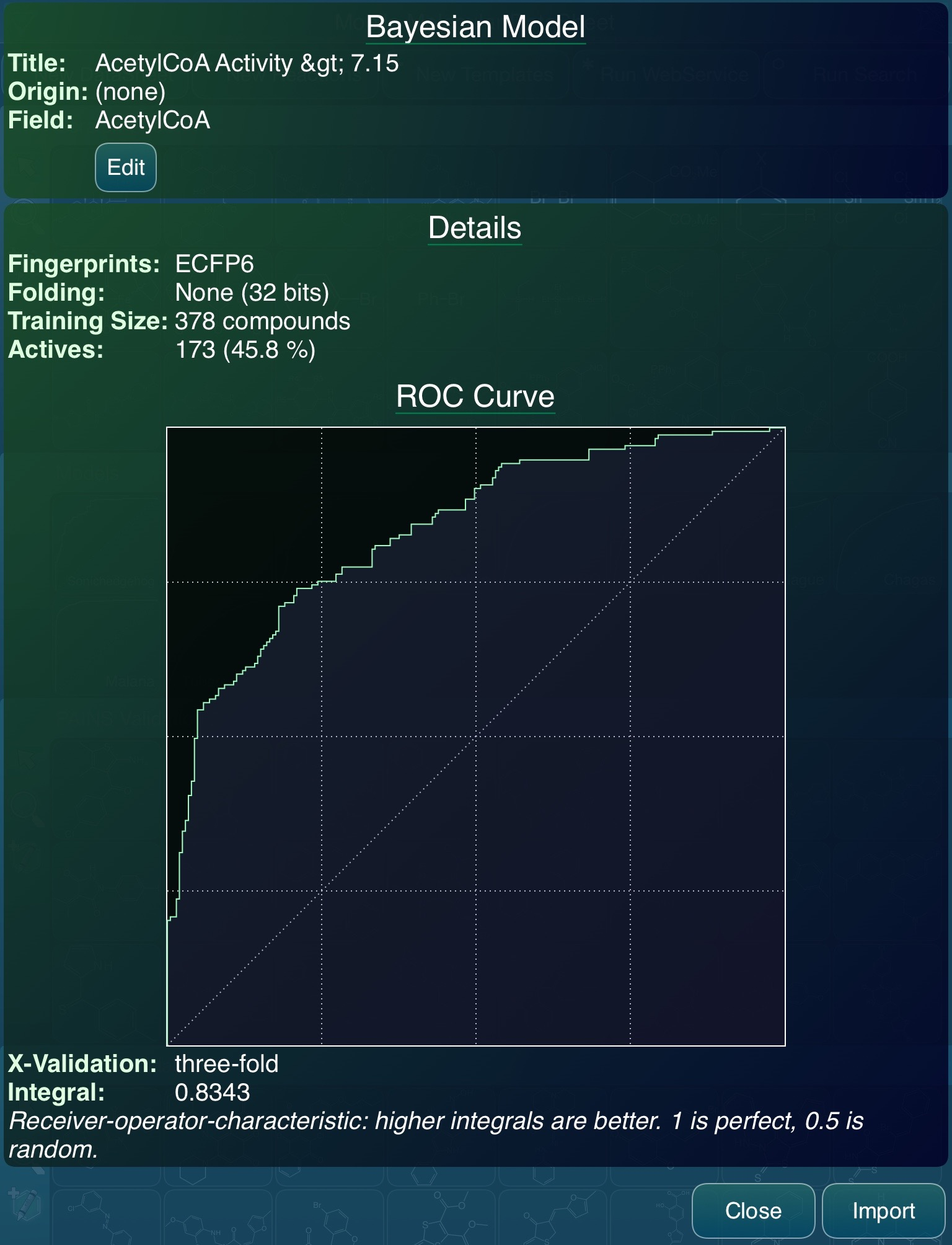
The import screen is informing us that yes, indeed, the incoming model was able to be parsed successfully. Note that it is slightly different to the one shown in Vault, because MMDS only implements the ECFP6 fingerprints, whereas Vault uses FCFP6, and the export process allows either to be selected. They are very similar in performance for most datasets.
 Once the model is imported, it’s yours. It joins the ranks of the list of models that MMDS has available to it, and will be there until you delete it. You can use it wherever you are, for as long as your iThing has battery power: no connection to the internet is required, because the algorithms necessary to apply the models have been built into the app itself.
Once the model is imported, it’s yours. It joins the ranks of the list of models that MMDS has available to it, and will be there until you delete it. You can use it wherever you are, for as long as your iThing has battery power: no connection to the internet is required, because the algorithms necessary to apply the models have been built into the app itself.
To see it in action, draw or select a compound, and invoke the Property Calculation view. This will carry out a number of calculations, and this list of functionality includes running the structure through all of the Bayesian models that are currently stored on the device, and for each one, producing a prediction number (calibrated to a probability-like scale) and a graphical overlay of which parts of the molecule contribute positively or negatively toward this prediction:

The MMDS app uses Bayesian models in this way on a one at a time basis, but it can also be used to populate a whole datasheet with predictions, i.e. adding new columns and populating them with numeric values.
All of these developments are very exciting, because the ability to share computational models is a really big deal, and has been holding the industry back for some time. Until now the contributions we have been making toward solving this problem were somewhat academic to a lot of people, e.g. if you’re not a programmer, having new functionality added to an open source toolkit may not help you very much. But now there are production tools that are available right now – for a very reasonable price – that can be used to create, share and consume models, which can be used with the peace of mind that if ever you want to use any of the proprietary products, there are open source analogs of everything you need to make it happen.
