 Cheminformatics for inorganic/organometallic compounds is the perennial afterthought that oft appears in the future work section, but never seems to get implemented: exotic bond types can be drawn in a way that is pleasing to other inorganic chemists but meaningless to computers, or in some cases vice versa, but never both. This article explores an approach to achieving such harmony.
Cheminformatics for inorganic/organometallic compounds is the perennial afterthought that oft appears in the future work section, but never seems to get implemented: exotic bond types can be drawn in a way that is pleasing to other inorganic chemists but meaningless to computers, or in some cases vice versa, but never both. This article explores an approach to achieving such harmony.
The example shown above features a cyclopentadienyl ligand, which has the same bonding arrangement as the archtypical ferrocene molecule. This is the 5-membered ring at the top, which is connected to the metal by all 5 atoms. This kind of bond poses quite a lot of difficulty to anyone trying to design a minimalistic representation for molecules that uses a bond graph. An attempt to represent the molecule in a way that captures all of the strong bonding connections must necessarily draw out all of these as explicit bonds (as shown in A), but this is ugly, and very different from the convention that chemists have settled upon, shown in B.
From a cheminformatics point of view, the representation A is quite good: all bonds are described; because cyclopentadienyl is a monoanionic ligand, just one of the carbon atoms is drawn as having a full single bond to the metal, while the remaining carbon atoms are connected to the metal with a zero-order (dotted) bond. This allows the valence calculations on the metal and ligand atoms to obey their normal counting rules, which is definitely a good thing. Besides aesthetics, the biggest problem with this representation is that it does not capture the symmetry. This could be achieved using fractional bond orders, but this approach introduces a slew of new problems (there is a very strong argument to be made for sticking with integral bond orders).
The two deficiencies – aesthetics and symmetry – can be dealt with by starting with the minimalistic representation of A, which is very easy to work from an algorithm point of view, and introducing an extra layer of metadata that indicates that this bond should have the rendering and symmetry characteristics that are shown in B.
This approach, which is still in its early stages, is being provisionally referred to as bond artifacts. The idea is to take a correct-but-ugly [and possibly insufficiently symmetric] representation and make it look the way chemists want to see it, while keeping the extreme minimalistic simplicity of a stripped down connection table with very few options.
There are three different types of bond artifacts, which I believe may be sufficient to cover an incredibly large proportion of resonance states and weird inorganic bonding modes:
- resonance paths
- resonance rings
- arene ligands
The following examples demonstrate several bond representation issues that can be addressed using resonance paths:
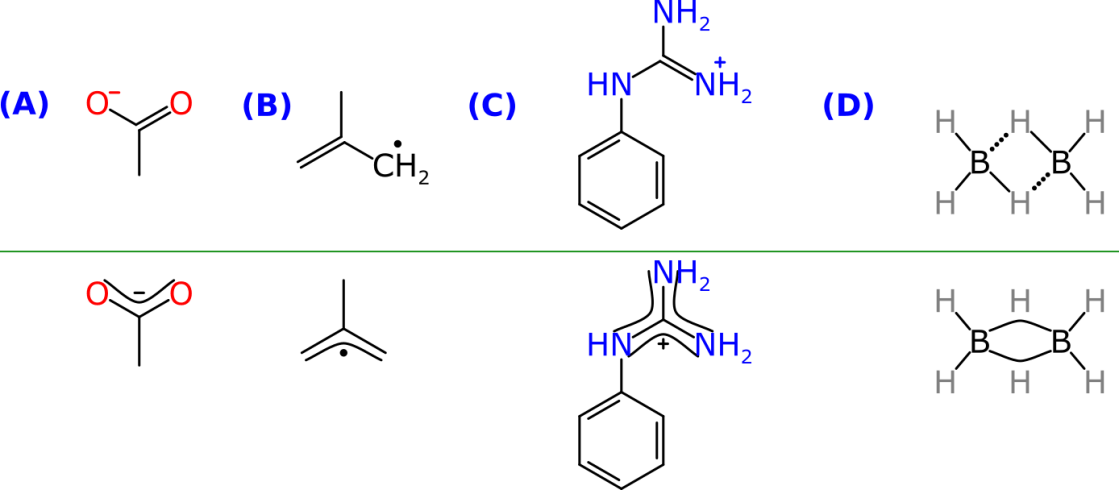
The structures above the line show the raw representation, i.e. one valid way to draw each of these compounds using integral bond orders from 0 through 3, integral charges, and a simple connection graph, such that the valences add up to the most chemically valid number.
Examples A and B are two very common and simple examples where the presence of a charge (carboxylate) or a radical (allyl) next to a double bond makes the affected terminal atoms completely equivalent due to resonance. In these cases there are two equally valid representations. Without the symmetry, though, it would really matter which of the two resonance forms was drawn, because cheminformatics methods would have to resort to fancy trickery in order to discover this equivalence (and as everyone ought to know, the thing about trickery is that no matter how clever you are, you’ll never get everything right). By introducing a resonance path artifact within the metadata, this equivalence can be formalised within the molecular representation, and provide the rendering engine an optional hint that it would be ideal to draw this resonance path using a style that is familiar to chemists, such as shown below the line.
These paths can also be used to solve more complex problems, such as the delocalised positive charge on the guanidine functional group shown in C. One way to solve the equivalence is to draw it as a carbocation, but this tends to be disfavoured by chemists, meaning that there can be up to 3 valid representations involving a charged imine, which can be tricky to disambiguate. The symmetry can be captured by adding 3 resonance pathways to the 4-atom functional group, which completely covers all of the bond delocalisation patterns. While the rendering shown above may not be the most beautiful way to draw this molecule, it certainly does get the point across.
The last example in the diagram shows the borane molecule, which features “3-center, 2-electron” (3c2e) bonding by hydrogen, which is a common recurring theme throughout inorganic chemistry. The low level representation that I recommend is to represent the 3c2e entity as a single bond and a zero-order bond, but most chemists would reject this immediately as a diagram option. However, by overlaying resonance paths over each of these 3-atom sequences, the rendering engine can hijack the bond drawing mechanism, and produce it as a single curve, which communicates the notion of it being a distinct bond, with corresponding symmetry. Meanwhile the actual connectivity is buried safe and sound within the raw bond graph.
Closely related is the issue of resonance rings, which may be more of a bane to regular organic chemistry with ordinary octet-abiding bond patterns, e.g.
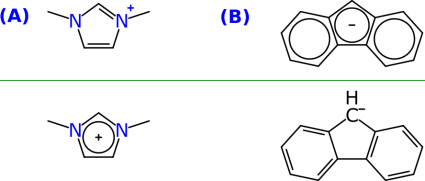
The concept resonance within a ring system is often confused with aromaticity, which is one of those unfortunate circumstances where the right answer is obtained for the wrong reasons sufficiently often that it’s hard to argue in favour of throwing it out. Rings that are obviously aromatic (e.g. benzene, pyridine, naphthalene, etc.) can be algorithmically detected easily, and likewise by chemists, who mostly prefer this to be implied within diagram representations. However, such algorithms can get into trouble if they are too aggressive about detecting “aromaticity”, but being insufficiently so is also problematic. Consider the imidazolium cation derivative in A: this 5-membered ring really is quite aromatic from a delocalisation point of view, and if the two substituents were different, then there would be two localised representations that seem different, but really refer to the same thing. The example in B shows the fluorenyl anion, marked up as fully delocalised. One argument for having an explicit bond artifact (rather than polishing up the detection algorithm for aromatic-like ring resonance) is that it is sometimes also advantageous to actually draw the resonance. Most chemists prefer their aromatic rings to be rendered in Kekulé form, i.e. alternating single/double bonds, rather than the ring notation. However, the fact that imidazolium has this property is more in need of explicit emphasis than for regular 6-membered rings, and there is a case for adding a command to render it so. A similar argument could be made for the fluorenyl anion, though whether it is obvious is a matter of opinion. And keep in mind that there are many oddball ring systems that have a lot of resonance delocalisation, but aren’t necessarily aromatic by a reasonable application of the Hückel rule, but they are nonetheless sufficiently delocalised that the bonds are interchangeable under normal conditions.
Finally we come to the arene ligands, which are a largely an organometallic phenomenon:
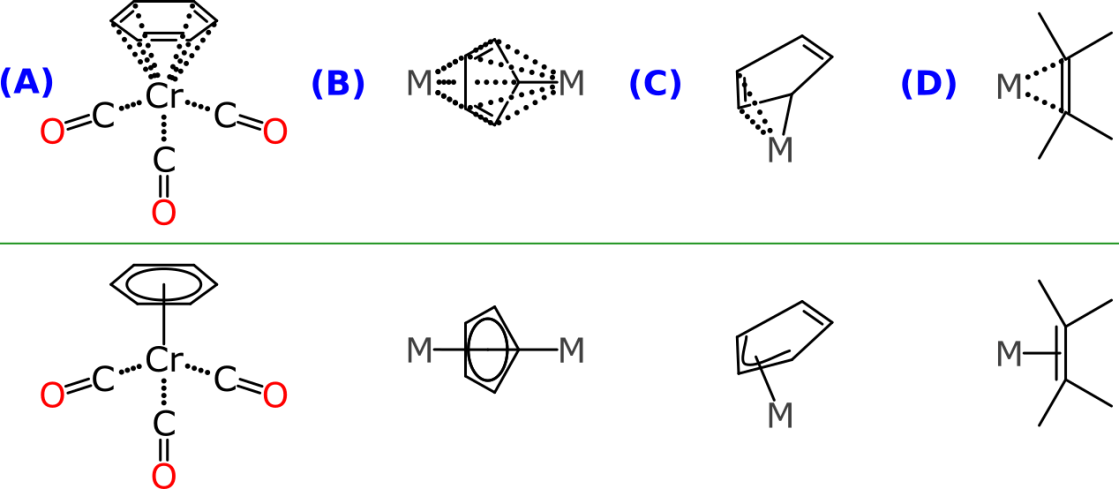
The datastructure to be layered over the core bond graph consists of a special central atom (typically a metal) and a set of atoms, which makes up either a path or a ring, depending on what sort of attachment is involved. Example A is another kind of arene, related to the cyclopentadienyl ligand found in ferrocene, except that it is a neutral 6-membered ring (coordinated benzene). Example B shows a fragment where a cyclopentadienyl ring is coordinated to two separate metals (yes, that is a thing). Example C shows another variation, where an arene is only partially bound to a metal (which is also a thing). Example B shows use of the datastructure to represent a bond-coordinated alkene, which is not technically an arene, but it follows the same principle.
The reason for showing these examples is to emphasise that inorganic chemistry has a lot of different bond types, and so the task of coming up with a canonical set of datastructures for each and every variant would be quite tedious, and inevitably incomplete. The approach that I’m presently experimenting with tries to make these as general as possible, and keep the incremental functionality as minimalistic as possible.
From a representation point of view, these are being encoded as optional extensions to the SketchEl datastructure. For example, the acetic acid salt captures the carboxylate group like so:
SketchEl!(4,3)
C=0.0000,0.0000;0,0,i0,xRESPATH:1:2
O=1.2990,0.7500;0,0,i0,xRESPATH:1:1
O=-1.2990,0.7500;-1,0,i0,xRESPATH:1:3
C=-0.0000,-1.5000;0,0,i3
1-2=2,0
1-3=1,0
1-4=1,0
!End
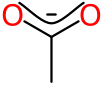
The first 3 atoms have the suffix xRESPATH, which is an optional extension to the atom. The two numbers indicate the group and the index within the group. Optionality means that it can be ignored without major loss of functionality, so not every software package that claims to be able to use the SketchEl format has to know or care about it; it just has to follow the rules and preserve the property during the read/modify/write cycle, even if it is not understood. This is an incredibly important characteristic that is missing from most (or all?) cheminformatics formats.
Rings are encoded in a similar way (with xRESRING), while arenes are slightly more nuanced: the first atom is special, being defined as the centre. So, ferrocene is represented as:
SketchEl!(11,20)
Fe=0.0000,0.0000;0,0,i0,xARENE:1:1,xARENE:2:1
C=-0.0154,1.0746;0,0,i1,xARENE:1:2
C=-1.0352,2.4083;0,0,i1,xARENE:1:3
C=-1.5164,1.4890;0,0,i1,xARENE:1:4
C=1.5028,1.4200;0,0,i1,xARENE:1:5
C=1.0642,2.3603;0,0,i1,xARENE:1:6
C=0.0051,-1.0747;0,0,i1,xARENE:2:2
C=1.0121,-2.4181;0,0,i1,xARENE:2:3
C=1.5020,-1.5035;0,0,i1,xARENE:2:4
C=-1.5164,-1.4055;0,0,i1,xARENE:2:5
C=-1.0868,-2.3500;0,0,i1,xARENE:2:6
3-4=2,0
4-2=1,0
2-5=1,0
5-6=2,0
6-3=1,0
1-2=1,0
4-1=0,0
5-1=0,0
6-1=0,0
3-1=0,0
8-9=2,0
9-7=1,0
7-10=1,0
10-11=2,0
11-8=1,0
1-7=1,0
9-1=0,0
10-1=0,0
11-1=0,0
8-1=0,0
!End

Note the two separate blocks using the xARENE extension.
At the present time, these extensions are coded up within the back-end Java library used by Molecular Materials Informatics, and interactively within the XMDS Mac desktop cheminformatics editor. There will be more information about that later: XMDS is on the Mac App Store, but the version that supports interactive creation and viewing of bond artifacts will be ready for submission soon. And at some point, it will be added to the WebMolKit open source library.
