 The most recent version of XMDS has a couple of useful new features: creating an abbreviation out of a group of atoms automatically reuses known labels, and there’s now an RDfile export feature for datasheets with the reaction Experiment aspect.
The most recent version of XMDS has a couple of useful new features: creating an abbreviation out of a group of atoms automatically reuses known labels, and there’s now an RDfile export feature for datasheets with the reaction Experiment aspect.
Well defined inline abbreviations have been a part of the XMDS project from the beginning, as well as the SketchEl molecule format and various mobile apps such as MMDS. The idea of an inline abbreviation is that it is represented by a single placeholder atom, but its internal definition is actually a fragment, and this fragment is encoded within the structure definition itself. This is as opposed to the usual ad hoc method of assuming that pseudoatom names like Me, Et, Ph, etc. will be recognised and handled properly (which I can summarise in two characters: no).
Use of abbreviations would be a lot less complicated if they were just strings text, but it’s not that simple. Consider two of the most common:
- nitro: NO2
- tertiary butyl: tBu
Note the subscript and superscript. From a typography point of view, that’s even more tricky than it looks: for the first case, you would expect the N of the nitro group to be at the “centre” of the atom (just as you would with, say, —NH2), and similarly for the butyl substituent, you would not expect the superscripted “t” to be the major layout component, rather it would stay out of the way.
Additionally, for the nitro group, you would expect it to be able to render backwards when it makes more sense to do so, i.e. O2N— (just as you would for H2N—), which means that more metadata is needed so that the renderer can decide how to reverse the components in the label.
For this purpose, element labels in the SketchEl format have some extra features:
- components are separated by “|”
- subscripts are surrounded by curly braces, i.e. {thing}
- superscripts are like subscripts plus a caret, i.e. {^thing}
So, the nitro group is represented as “N|O{2}” and the tertiary butyl group is “{^t}Bu”. The tertiary butyl group doesn’t have a reverse representation, because it is just one component, but the nitro group can be switched around to “O{2}|N”, with the N component being the central position.
This metadata format is not the most complex ever invented, but it also isn’t very intuitive, and there isn’t any super convenient documentation or contextual clues in the user interface of XMDS. That being said, the abbreviations can be created automatically with their proper labels by using the templates, which include a number of substituents that can be added as either whole fragments or their abbreviated version. The most recently added feature affects the post hoc creation of abbrevations, i.e. you’ve already drawn the fragment, then you want to subsume it into an abbreviation because it’s too unwieldy.
This is better represented in pictures, so here’s an example molecule being edited with XMDS without any abbreviations yet:
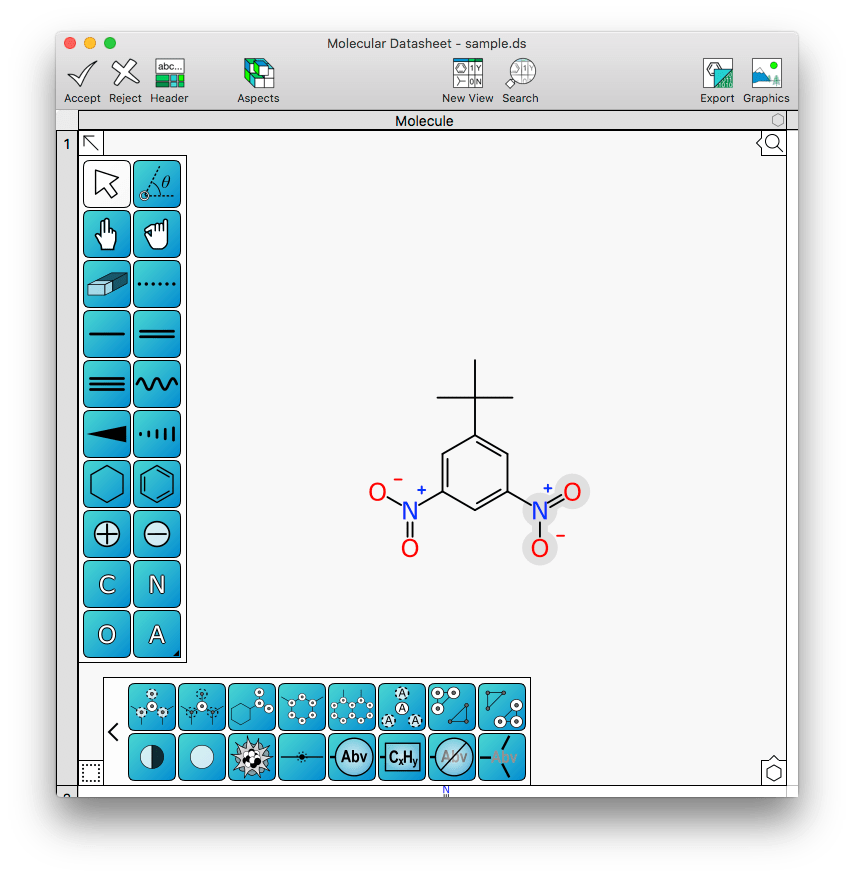
Shown above, the rightmost nitro group has been selected. Hitting the abbreviation icon in the command bank (or right clicking and selecting Abbreviate Group) will turn this external fragment into an inline abbreviation:
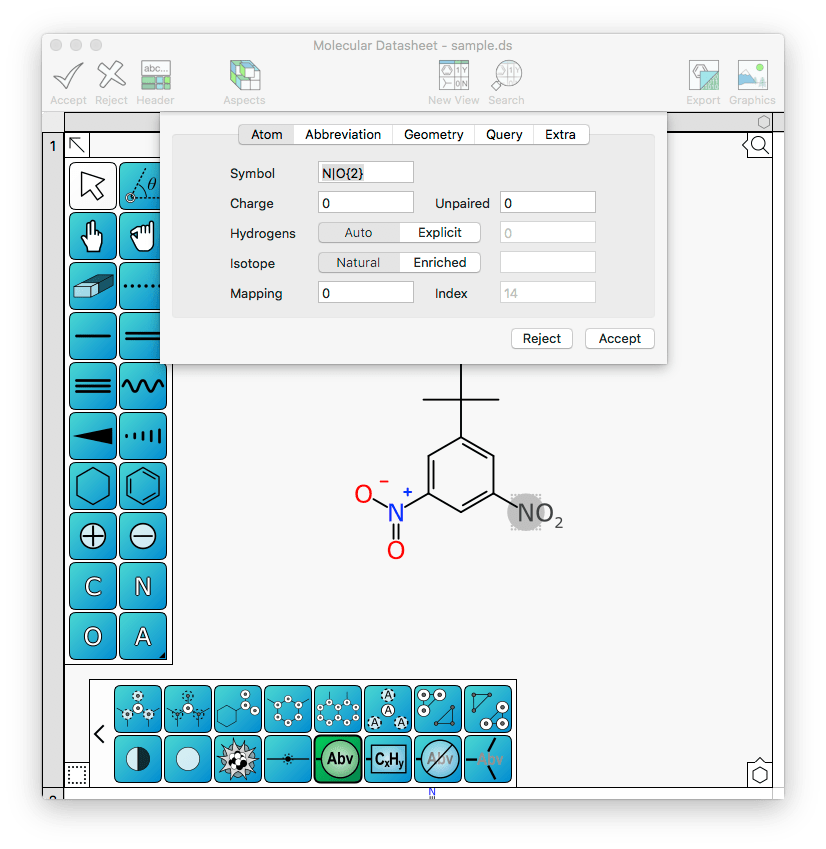
The previous behaviour was to assign the name of the abbreviation to “?” and let you type in what you want it to be. Now, however, it does something a bit more clever: it scans through its list of known abbreviations, and checks to see if any of them have the same connection table. If they do, then that abbreviation name is pulled out and used. As you can see in the above example, the definition is immediately set to “N|O{2}” (and is displayed in the molecule as NO2). It is also nicely lined up, with the direction of the bond pointing to the very middle of the “N” glyph.
Now likewise for the tertiary butyl fragment at the top of the ring:
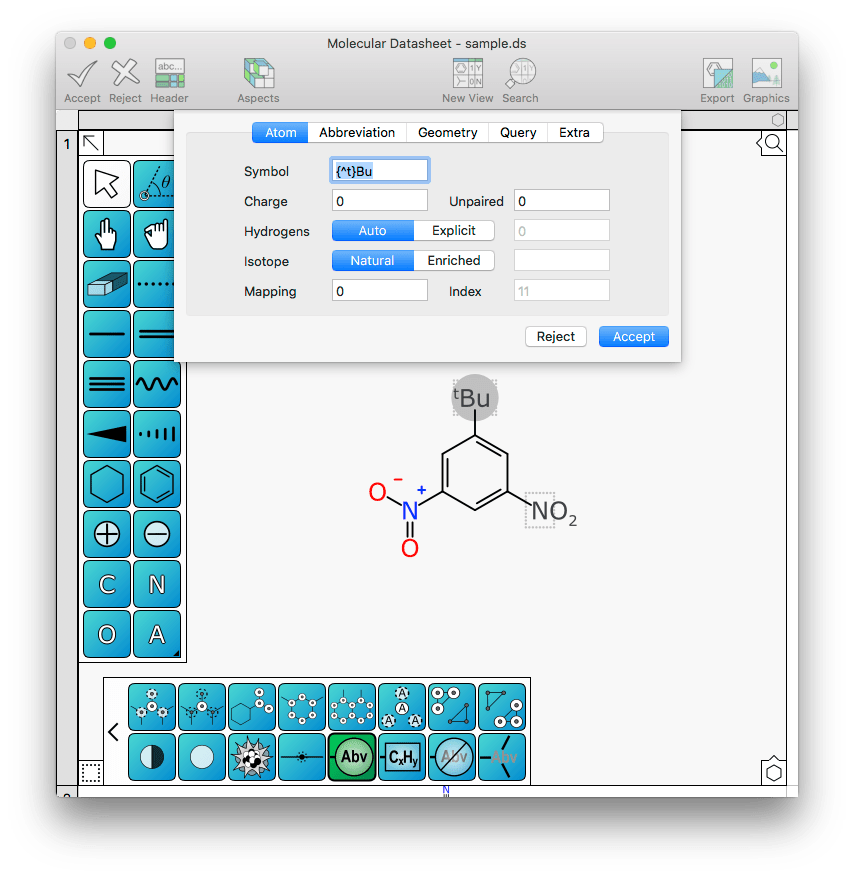
Now there’s no need to remember how to describe the “{^t}Bu” label, because it automatically finds it from the templates.
Note that it’s not just templates that furnish the abbreviations: the app also scours any of the files you load and scrapes the abbreviations that you’ve been using, so when you make up your own, you might just see it appear automatically at a later date.
This ability to remember abbreviations (from templates and elsewhere) is also evident when you switch to the Abbreviations tab:
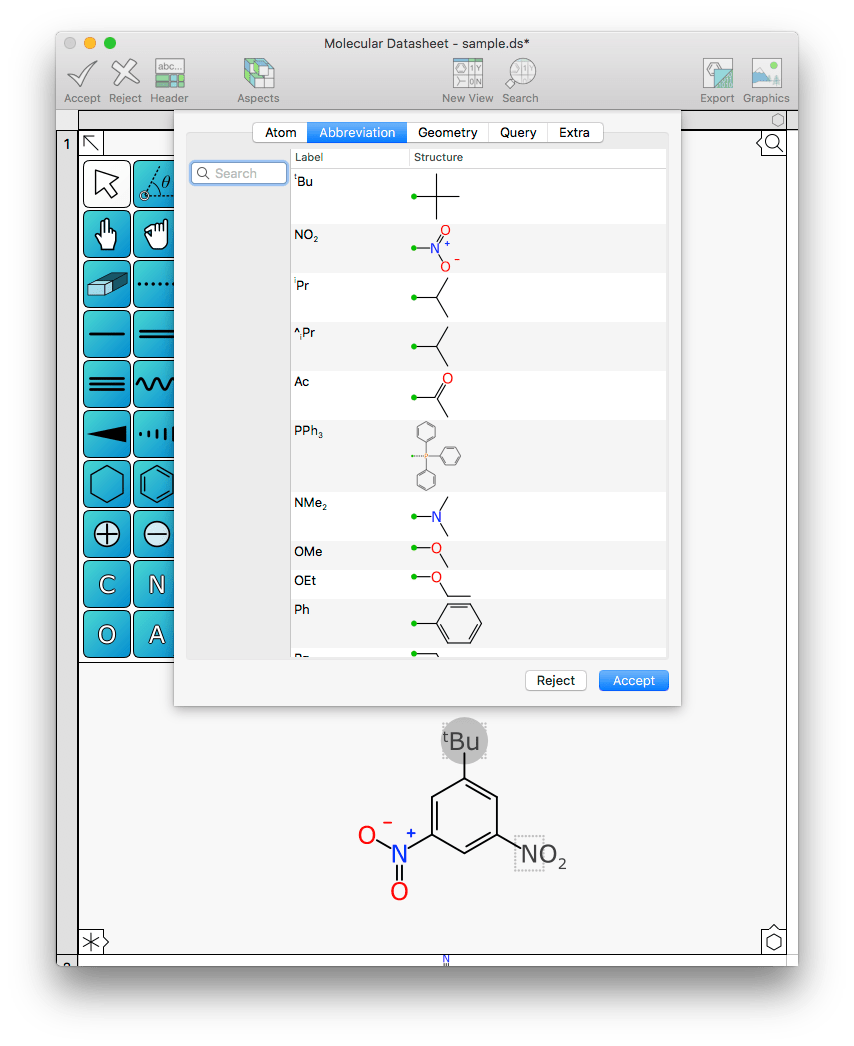
The list can be searched by typing in partial names, and selecting any of them overrides both the label and the fragment. (And if you look closely you’ll see there’s an isopropyl fragment that uses the wrong syntax for the label – this was from the current datasheet, which contains a mistake.)
Back to the last fragment needing to be abbreviated, and note how the leftmost nitro group is rendered backwards:
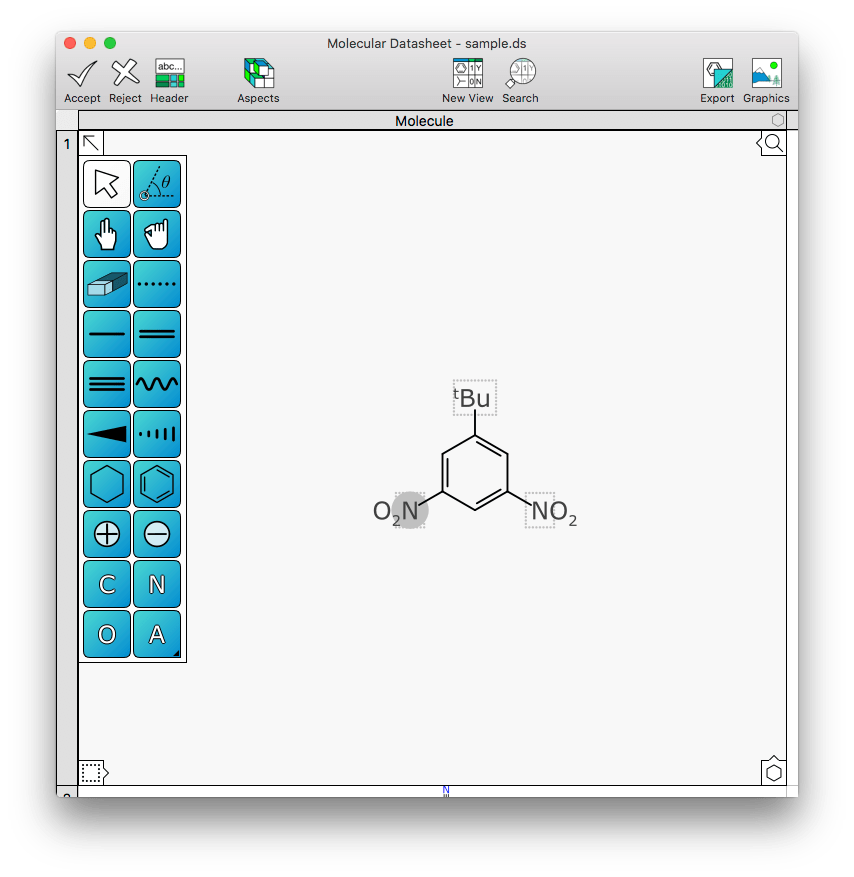
The other feature that has recently been added is the ability to export datasheets that contain the Experiment aspect to a file format other than its own, namely RDfile. The RDfile format was originally established by MDL a long time ago, and like the other formats from this batch, it is quite horrible for a long list of reasons. Unlike Molfile and SDfile, it does not have a vast audience of software for reading & writing, but it is about as close as we have for a standard for chemical reactions. It is different from the more common approach of representing all reaction components with one large canvas (i.e. what most people do with ChemDraw), and it can be used to store multiple reactions, each with their components broken down into individual Molfiles. In this regard, it is an effort to represent chemical reactions in a cheminformatics-friendly way.
For most of its life, the RDfile format has only been able to define two different types of reaction components: reactants and products. This means that anything that a chemist would have drawn above or below the reactant line (i.e. reagents) either has to be upgraded to a reactant, or represented without a structure, using just text. This basically makes the format more or less useless for serving up a reaction in a form that chemists would like to see. However, I happened to hear a rumour at a meeting last summer that there was a general consensus about the industry that the missing 3rd component type is recognised by any self-respecting software, and so it should be quite safe to use. This opens the door for exporting the kinds of reactions described by XMDS, MMDS or GLN (all of which use datasheet aspects) in a way that’s not completely lossy.
Consider the following reaction collection:
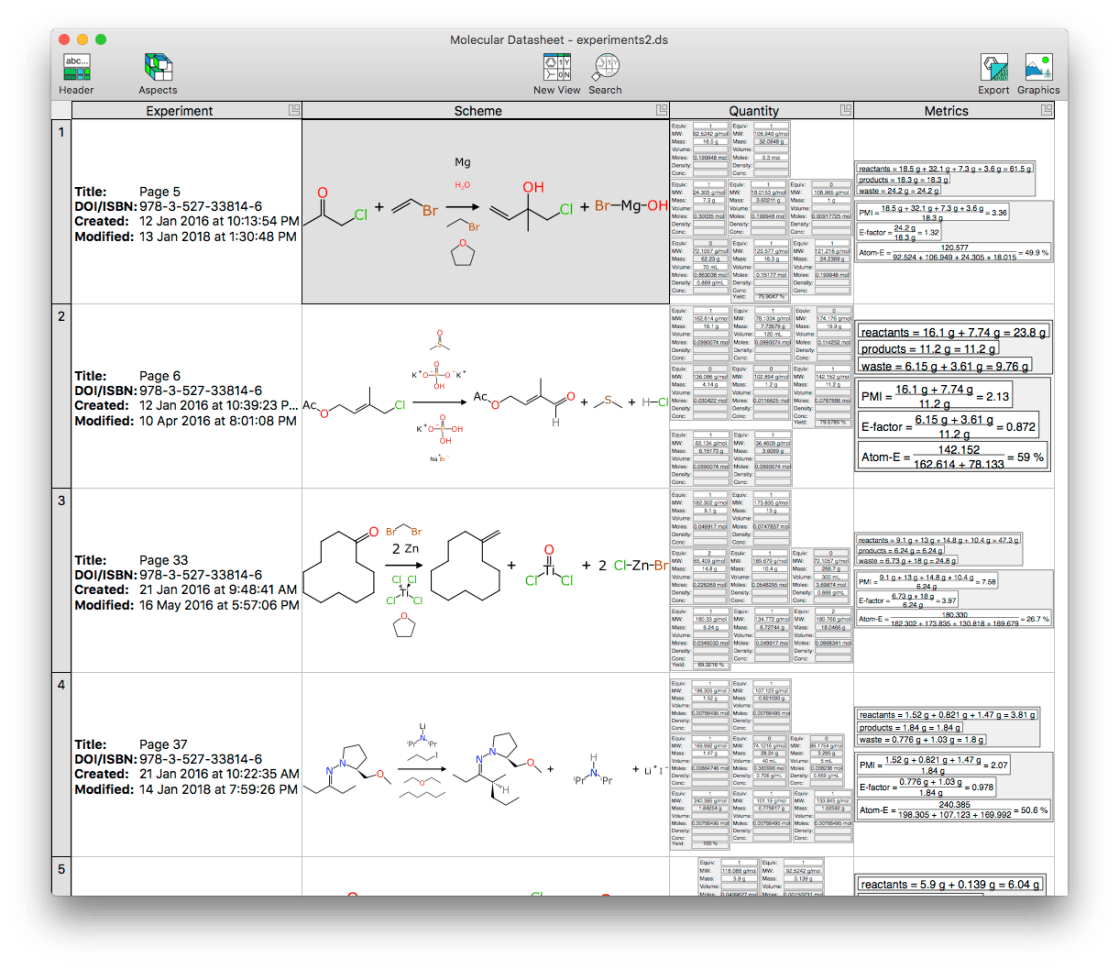
The Export dialog now has an option for RDfiles, allowing them to be offloaded to files, clipboard or drag’n’drop:
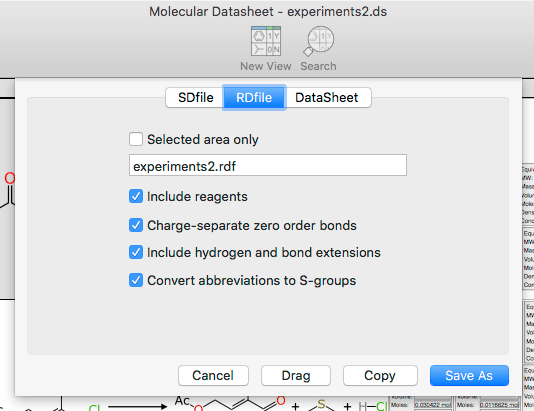
There are several options, such as the option to not include reagents (for compatibility with un-augmented readers), and the various finagling that has to be done with the constituent Molfiles to guarantee compatibility when the functionality gets a bit more interesting.
As a sanity check, the RDF importing (with reagents) works with the latest importer from Marvin/ChemAxon, which is generally more authoritative than reading the official specification (since the format is semi-abandoned). It works:
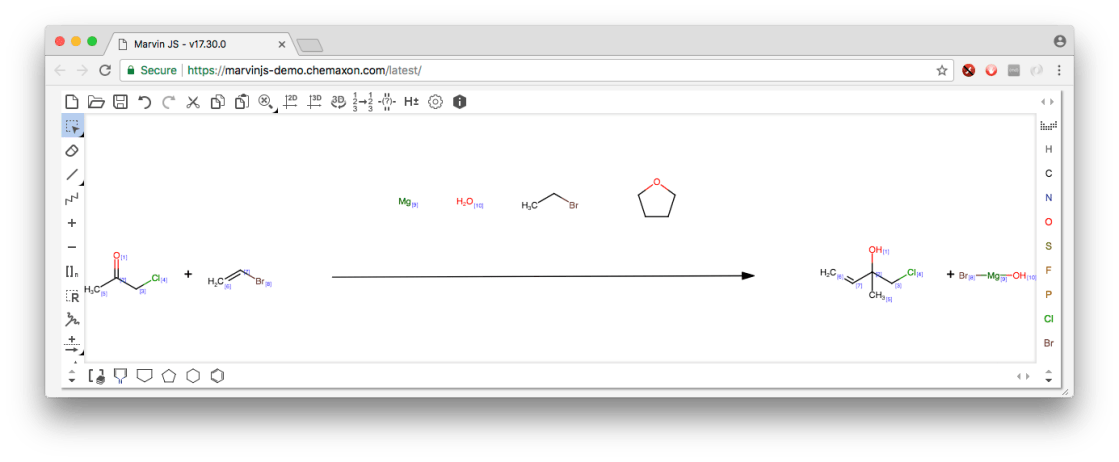
At the moment XMDS is not able to import RDfiles, but following up with that is high on the to-do list. It may be possible to have most of the metadata survive a round trip, which would be an interesting development.
These two features are available in the current beta release (which is still open to anyone who is willing to step forward and ask nicely), and will be incorporated into the next AppStore version.
