Two new features have been added to the XMDS macOS app lately (see beta & AppStore): fine control of atom/bond geometry from a dialog box, and automatic display of atom oxidation state.
Geometry control by entering numbers into a dialog box is a feature that should not be necessary in a chemical sketcher very often, but every once in awhile it’s the best way to get the desired outcome. For almost all routine operations, the new atom/bond placement tools employ logic for calculating the ideal position, and in most of the cases where they do not, operations such as rotate/grow/shrink/flip provide convenient corrections. Positioning atoms by freeform mouse dragging will do in a pinch, but eyeballing it does not always result in a great sketch, especially if there is symmetry involved (i.e. going freestyle for one atom might work, but getting the same result for two is much harder). Sometimes it’s better to enter angle & distance, and go straight to the ideal answer.
One example that comes up quite often is when starting with a 5-membered ring, which happens to have a long zig-zagged alkane branch. Ideally the ring would be platonic and align with at least one axis, but also the alkane bonds should be ±30° to one of the axes, too.
What usually results from the initial sketch is a structure like this one:
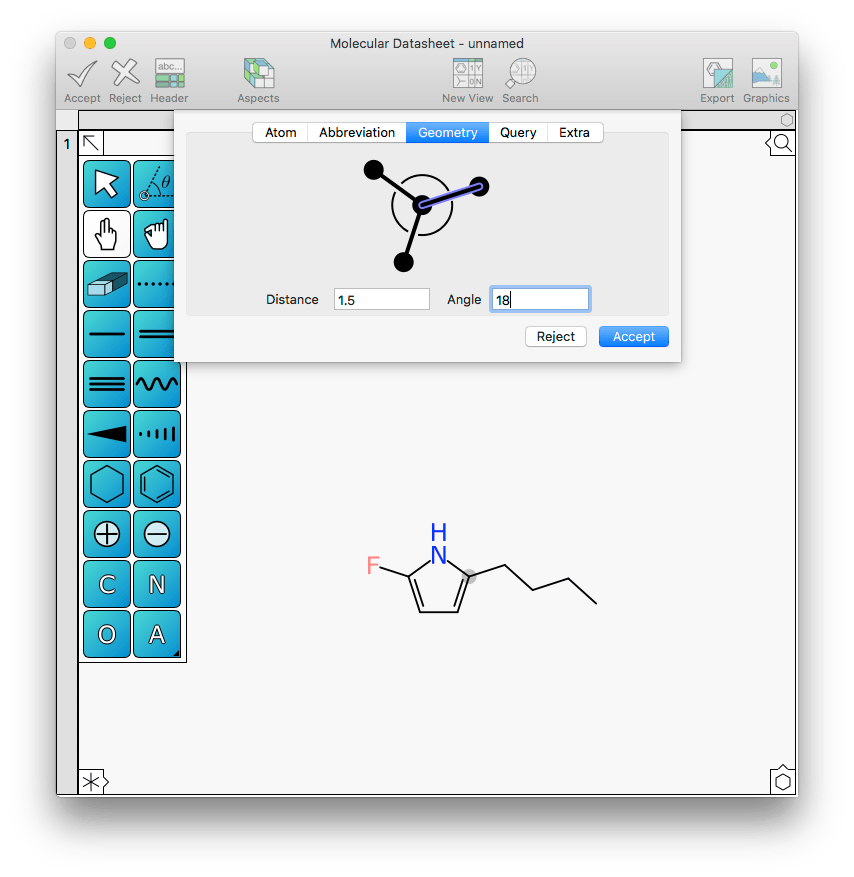
In this case the ring is ideal, but the sidechain is not: it would be nice if it aligned with the X-axis. Rotating the alkane sidechain incrementally is one way, but another is to open the new Geometry tab within the atom editor menu and change the angle to 30:
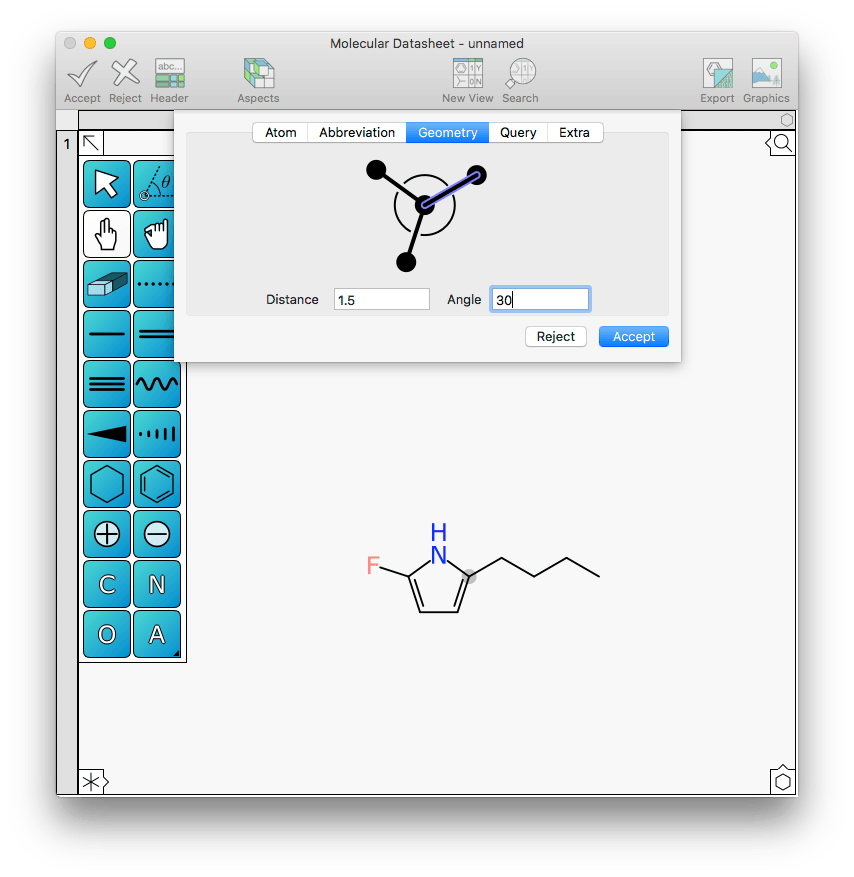
And in one operation the change is made, without any approximations or need for hand-eye coordination.
In the above example the absolute angle of the substituent was modified, but it is also possible to select the angle between two substituents to move them closer or further apart. For example, the two iPr abbreviations on this central atom might be considered a bit too congested:
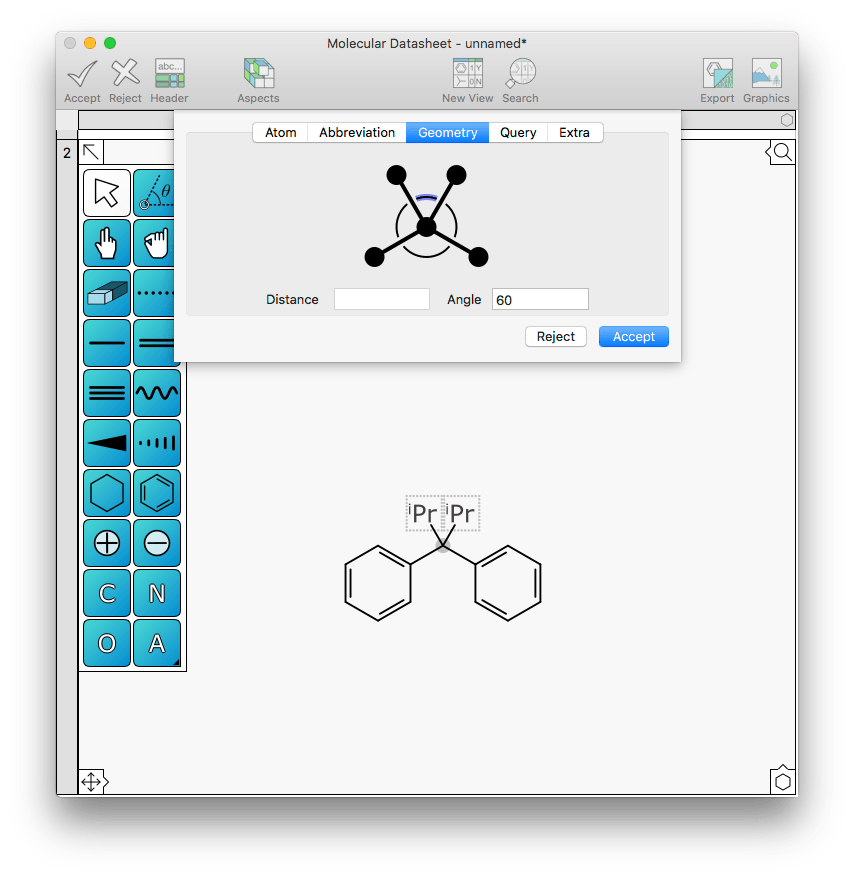
This is easily fixed by changing the angle between them from 60 to 90:
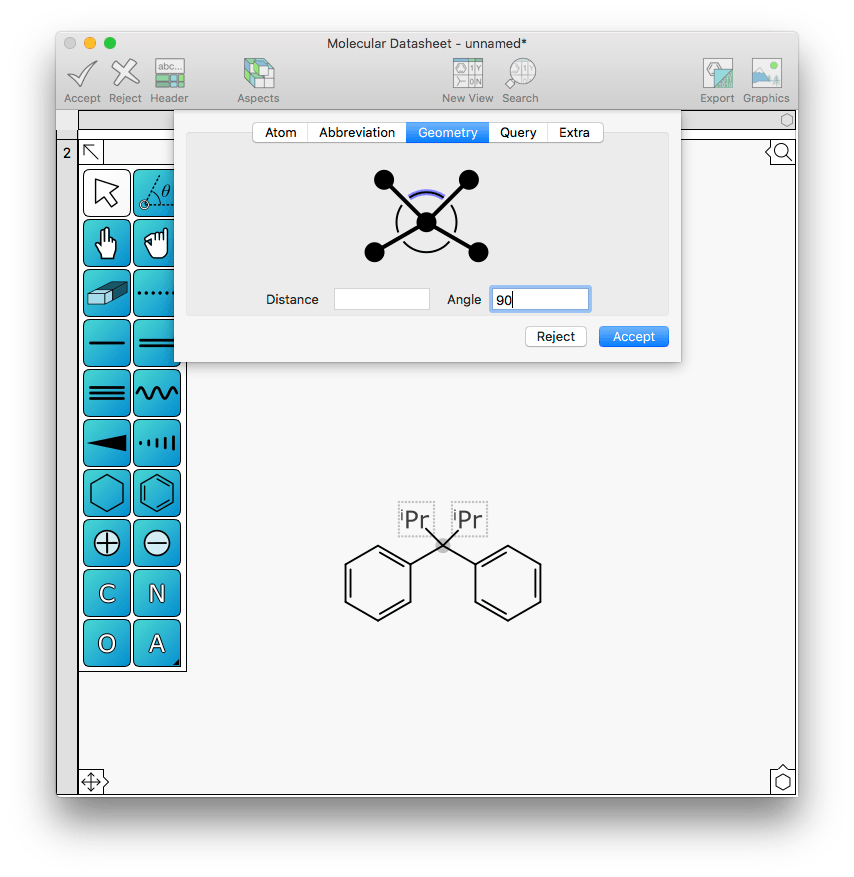
Similarly with bond distance: this can be achieved with the shrink and grow primitives, but it can now also be done by editing the bond and entering a new distance value. For example, the two triphenylphosphine ligands might be a bit too close to the rest of the metal action:
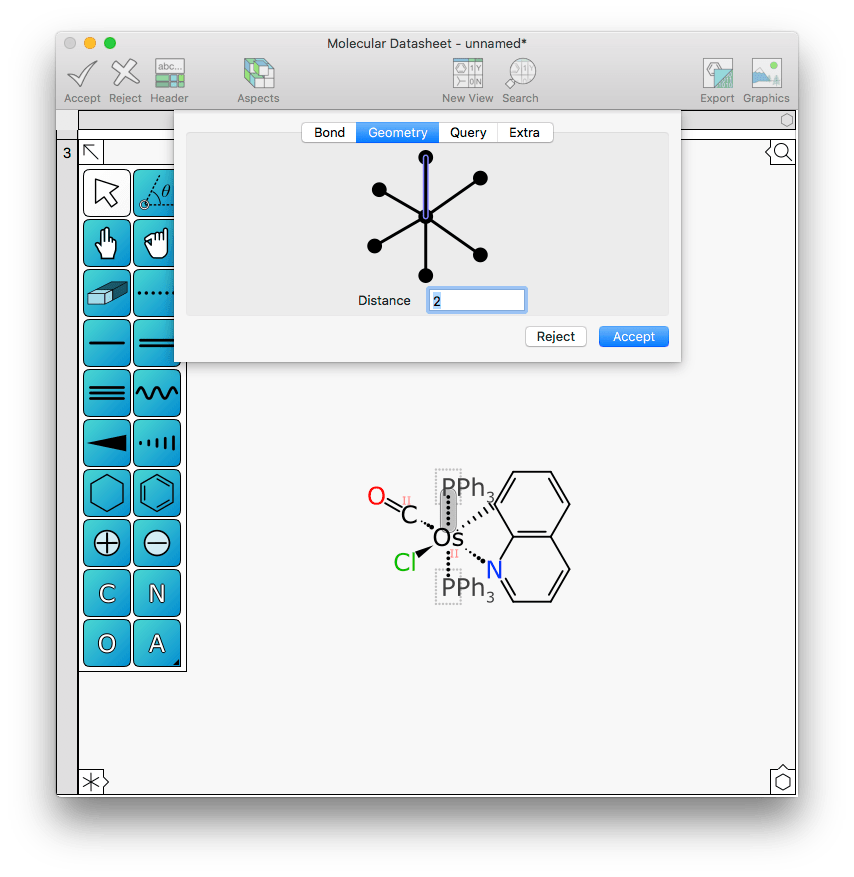
Changing the distance from 2 to 2.5 Å makes this a bit more clear:
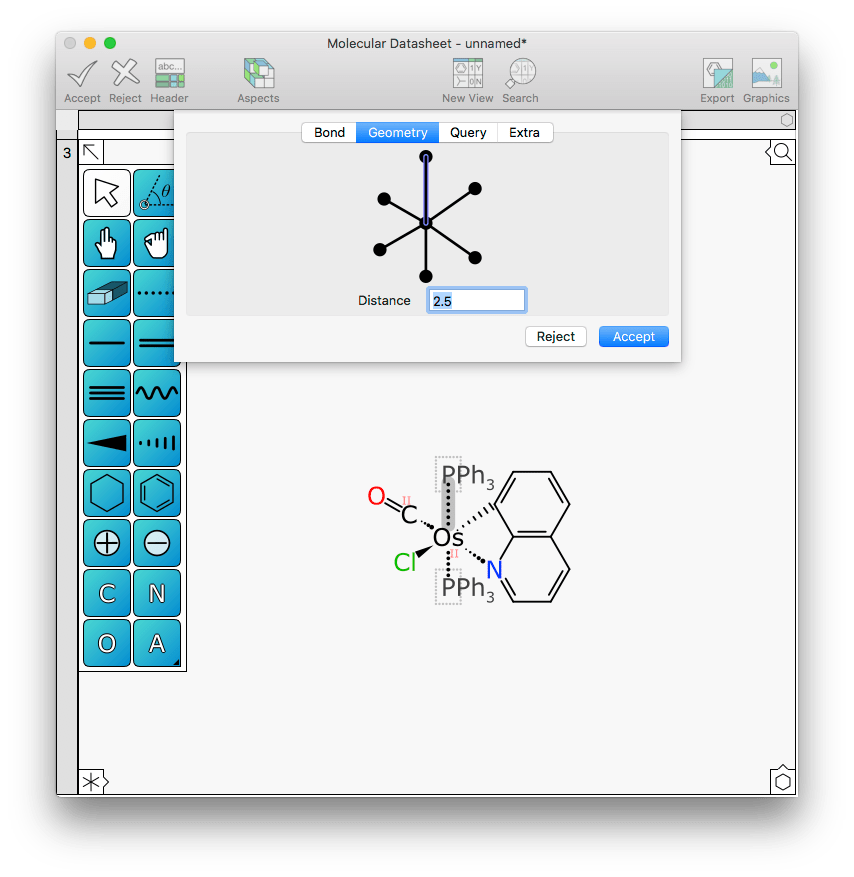
Note also in the above diagram, two of the atoms have an additional annotation that wasn’t present until the most recent version: the osmium centre is labelled with II on account of being in the +2 oxidation state, based on the way metals are assigned. Also, the carbon of the carbonyl ligand gets this label as well, because it is in a decidedly unusual state, being formally carbon II.
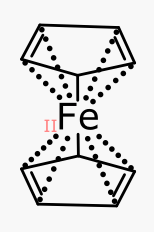
The display of oxidation state is only on by default while in edit mode, so it does not pollute any presentation graphics, similar to the default-on display of R/S/E/Z stereochemistry while editing. The feedback is useful: for metal coordination sites, getting the valency right often involves use of zero-order bonds, which are unfamiliar to most chemists, since such concepts have been largely left out of the realm of cheminformatics. The canonical problem child – ferrocene – can be drawn a number of ways, but the best ones are those for which the correct oxidation state is implied, e.g.
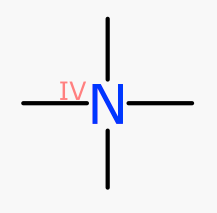
For regular organic molecules, oxidation state is only shown when there’s something unusual to see, and this is often indicative of a mistake. The classic tetravalent neutral amine is a common one:
Since it was released on the AppStore a few months ago, XMDS has continued to be refined for improved usability, and is evolving quickly. And of course for each headline feature that gets a blog post, there are usually a number of quiet bugfixes.
