 Several of the flagship apps from Molecular Materials Informatics have had major updates recently: the Mobile Molecular DataSheet, SAR Table, MolPrime+, Green Lab Notebook and Approved Drugs. Two separate groups of features have motivated these updates: (1) the inclusion of in-app calculation of nontrivial properties, lately supplemented by the inclusion of Bayesian models, and (2) leveraging the new iOS 8 API feature for importing & exporting data to any compatible service, which includes iCloud by default, but also Dropbox if it is installed.
Several of the flagship apps from Molecular Materials Informatics have had major updates recently: the Mobile Molecular DataSheet, SAR Table, MolPrime+, Green Lab Notebook and Approved Drugs. Two separate groups of features have motivated these updates: (1) the inclusion of in-app calculation of nontrivial properties, lately supplemented by the inclusion of Bayesian models, and (2) leveraging the new iOS 8 API feature for importing & exporting data to any compatible service, which includes iCloud by default, but also Dropbox if it is installed.
Each of these has been mentioned in a separate blog post or several: Bayesian models in Approved Drugs, PAINS filters in MolPrime+, advanced properties in MMDS and file import/export in GLN. As of now, a batch of additions have all been approved and are available on the iTunes AppStore, so here is a scorecard for that which has been added to what:
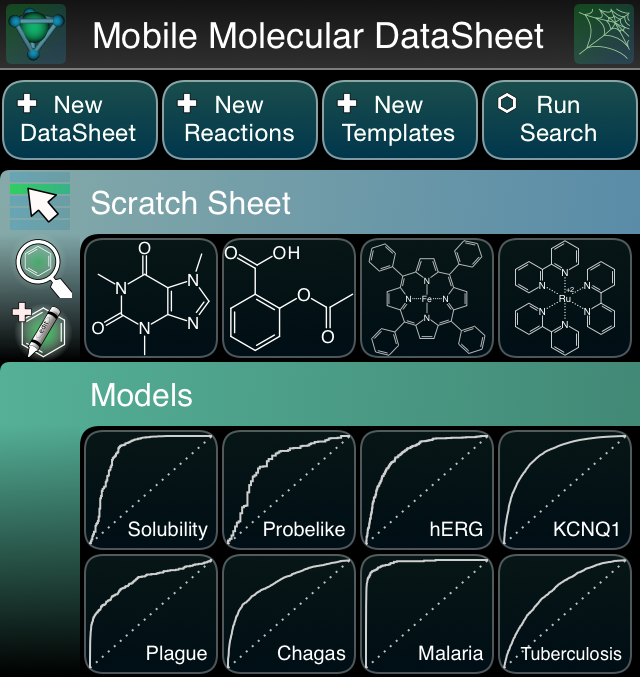
- Mobile Molecular DataSheet (MMDS): in-app property calculation: single molecule visualisation and calculation of whole columns for a table; 8 prepackaged Bayesian models, which are currently read-only, but will subsequently allow creation/editing/importing/exporting; importing & exporting of molecules and datasheets via iCloud mechanism
- SAR Table: importing & expecting of datasheets (Bayesian modelling will be coming later)
- Approved Drugs: 4 of the prepackaged Bayesian models (solubility, Lipinski probelikeness, hERG & KCNQ1 avoidance) with atom fragment visualisation
- MolPrime+: property calculation & prepackaged Bayesian models, for individual compounds
- Green Lab Notebook: importing & exporting of datasheets
The miscellaneous property calculation features are described in the official documentation, which includes a variety of scalar properties (from easy things like molecular weight to not-so-easy, like log P); stereochemistry; tautomers; valence violations and PAINS filters. These are all provided by algorithms that have been ported to Objective-C and reside within the app proper, so there is no longer any need to call out to webservices for the tricky stuff.
The Bayesian models are now documented: this is functionality that traces back to a joint project with Collaborative Drug Discovery to create an accessible and portable equivalent to the original SciTegic ECFP and FCFP fingerprints, which were submitted to and incorporated into the Chemical Development Kit (CDK) library (as Java code), and subsequently reimplemented in the MMDS core library (i.e. recreated in Objective-C). Since the fingerprints were designed and documented in such a way as to make it as easy as possible to reimplement exactly the same results on different cheminformatics platforms (as described in our paper), it means that anyone can rebuild the algorithm from thorough and step-by-step instructions, then use the CDK toolkit to generate validation examples to ensure that they really are the same. Or if your project is compatible with the Gnu Public License, and can use a Java Virtual Machine, you can just use CDK as-is.
More recently, working with Collaborative Drug Discovery once again, we released a class for creating, using & serialising Bayesian models, and submitted it to the CDK project. Over the last year and a half, I have reimplemented the basic Laplacian-corrected Bayesian algorithm about a dozen times in order to explore various different ideas, and the latest effort is quite robust, and now resides in the CDK codebase with a respectable set of validation tests. This is a recent development – you can find the package on the latest version on Github: look for the tools section, for class org.openscience.cdk.fingerprint.model.Bayesian.
This is important, because the CDD-sponsored CDK module can be used to build Bayesian models based on ECFP/FCFP fingerprints, and serialise them as portable model files. The file format that the MMDS, MolPrime+, Approved Drugs and soon SAR Table apps use within their prepackaged bundles is the same format that the CDK implementation can create & consume. Even though it’s a separate codebase, it has been tested on the same validation examples, so they completely compatible. All the gory details will be published in the literature in the near future. While the mobile apps currently do not have the ability to add or create models, that is going to change soon enough.
If Bayesian modelling of structure-activity data is interesting to you, keep an eye on new developments from apps from Molecular Materials Informatics, web informatics from CDD Vault, and open source tools and algorithms from the Chemical Development Kit. These platforms will soon have compelling new reasons to be used together.
