 Normally I wouldn’t consider a trivial dialog box to be worthy of a blog post, but it’s as good a time as any for a progress update on XMDS. The sketching interface for editing molecular structures is now somewhat feature complete. There are a few bits to come back to, but in general the methods for adding, deleting, editing and aligning atoms and bonds are available in a highly redundant way. By redundant, I mean that in a good way. The editor is based on a confluence of methodologies that I’ve built out in various products over the years: (1) the conventional way of drawing molecules with a painting-style toolbox (see SketchEl); (2) the “drawing primitives” designed for precise editing when user interaction is expensive, motivated by mobile devices with tiny screens; and (3) keyboard shortcuts, designed so that an expert can draw molecules incredibly quickly without having to reach for the mouse, battle-tested with the BlackBerry version of MMDS back in 2010, when touchscreens were not yet ubiquitous.
Normally I wouldn’t consider a trivial dialog box to be worthy of a blog post, but it’s as good a time as any for a progress update on XMDS. The sketching interface for editing molecular structures is now somewhat feature complete. There are a few bits to come back to, but in general the methods for adding, deleting, editing and aligning atoms and bonds are available in a highly redundant way. By redundant, I mean that in a good way. The editor is based on a confluence of methodologies that I’ve built out in various products over the years: (1) the conventional way of drawing molecules with a painting-style toolbox (see SketchEl); (2) the “drawing primitives” designed for precise editing when user interaction is expensive, motivated by mobile devices with tiny screens; and (3) keyboard shortcuts, designed so that an expert can draw molecules incredibly quickly without having to reach for the mouse, battle-tested with the BlackBerry version of MMDS back in 2010, when touchscreens were not yet ubiquitous.
One of the necessary ingredients for a keyboard-enabled method for editing molecules is the ability to select atoms and bonds using the cursor keys. The arrow keys can be used to walk the current atom/bond across the molecule, by hopping through connected atoms, or jumping across empty space where appropriate. This can be used in conjunction with actions like sprouting new bonds in their most promising locations (accessible from the keyboard, of course). For a desktop product, this is motivated by the fact that humans don’t have 3 hands, and since the keyboard is a 2-handed tool, the ergonomics of moving one of them to the mouse/trackpad can be considered an expensive operation. On the other hand, if figuring out what to do takes more than a couple of seconds, it doesn’t really matter, which means that there is a strong argument for supporting both: all hands on keyboard for “touch-typing” when you know what you want, one hand on the mouse for “hunt-and-peck” when being indecisive for whatever reason.
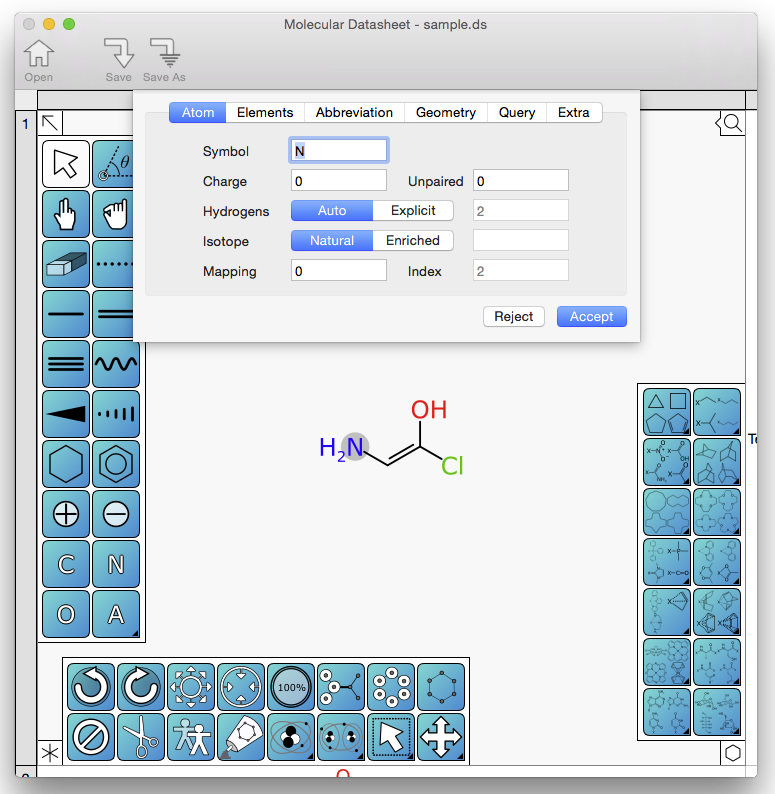
One of the 4 view panels that can be shown onscreen is the detail view which is displayed at the top, and reacts according to the selection, to show information about the whole molecule, or the particulars of an atom or bond. Originally I had intended to make it fill out editable fields and widgets so that they can be modified in the same panel without the jarring context switch of popping up a dialog box. After a bit of struggling against the gnarliness of dealing with tab focus issues using the Cocoa API, I decided to try a more conventional approach. As it turns out, the preferred way to do a dialog box on the Mac is what is internally described as a “modal sheet”, which is the panel that slides down out of the title bar, and blocks the rest of the interface until it is dismissed. Functionally it is the same as a dialog box, except it moves around with the parent window. I had never given it much thought as a principal feature of UX/UI design – it seemed a bit irrelevant – but in this case it really works. While editing a molecule, moving the atom/bond focus around with the cursor keys, hitting return brings up the editing panel. Using keyboard entry to alter values (e.g. element, charge, hydrogen status, etc.), the tab key to switch between field, the return again to accept the changes, is a method of working that flows very naturally. It’s one of those things that could be either awkward & annoying or effective & pleasant, depending on the details, and it always seems obvious in retrospect.
Next steps will involve a return to working on the spreadsheet-like interface for editing entire datasheets, which currently doesn’t do much except let the user view, select, scroll, and open the molecule editor. The general idea for the XMDS project is that the version 1.0 minimum viable product will be primarily focused on data entry of structure-activity content, with a frictionless onboarding experience for everyone who has used Excel & ChemDraw, and fast & perfect drawing for anyone who takes the time to learn the more advanced features. Streamlining the drawing molecules and entering property information will be the top priority, since most of the options out there are not really optimised for taking the pain out of this task. And since the universe of chemical data includes so much of this kind of content that has yet to be curated, this is a clear and present value proposition.
The fun stuff that will be added after that is a very long list indeed, but first things first.

I bet this will revolutionize DD using Macs. The beta version looks fantastic.