 One of the opinions (arguably of the educated variety) that I’ve been pushing for awhile now is the idea that when a model building or visualisation technique requires a user parameter in order to get the correct result, that is essentially an admission of partial failure. If the method really was so great, then it would be able to figure it out, because a parameter is an extra degree of freedom that the method has punted on. Now of course this is not a rule by any stretch of the imagination, and there are numerous exceptions, or grey areas between what’s a parameter and what’s an integral component of the source data. But sometimes a parameter really is just something that a method ought to know, but gives up and passes the burden on to the user – and that’s not necessarily a bad thing, as long as we admit it.
One of the opinions (arguably of the educated variety) that I’ve been pushing for awhile now is the idea that when a model building or visualisation technique requires a user parameter in order to get the correct result, that is essentially an admission of partial failure. If the method really was so great, then it would be able to figure it out, because a parameter is an extra degree of freedom that the method has punted on. Now of course this is not a rule by any stretch of the imagination, and there are numerous exceptions, or grey areas between what’s a parameter and what’s an integral component of the source data. But sometimes a parameter really is just something that a method ought to know, but gives up and passes the burden on to the user – and that’s not necessarily a bad thing, as long as we admit it.
Which is what I’m doing in this blog post. Awhile back when I started added Bayesian predictions into apps (starting with Approved Drugs, then MMDS), one of the features that I came up with was atom highlighting, whereby the molecular structures are coloured with the constituent atom contributions from the Bayesian model. This is possible because the models are based on ECFP6 fingerprints, and the Laplacian-modified naïve Bayesian model is a collection of linear contributions from each of these fingerprints: because the fingerprints can be mapped back to the atoms that instigated their presence, it is possible to add up all the applicable contributions for a molecule, and smear out the contribution values across the atoms.
What this means is that the outcome now has a probability-like prediction value (in the range 0..1 most of the time) for the whole molecule, and for each atom, it has a numeric value that can range between… well, anything to anything.
The intention of highlighting the molecular structure with atom-specific contributions is to show which parts of the molecule are particularly good or bad; for example, some molecules might be split in half, whereby one side contains fragments that always appear in actives and the other side has only inactive-type-stuff. But sometimes, all components of the molecular are pretty much the same. Deriving enough information to rank the atoms is easy enough, but the objective is to be able to tell the user that one region is much better than another, and that requires calibration. If the range between best and worst is 0.1, is that a lot? Or is it just noise?
I’ve put a fair bit of thought into this, and so far, I don’t really have a good answer. All of my ideas for figuring out how to use atom-smeared uncalibrated Bayesian contributions to make this judgement call involve circular reasoning, or some hare-brained scheme for running stats over a large database using some contrived scoring function.
Until I had a minor epiphany, and decided that because I don’t have a good answer right now, so I’m just going to punt and make the calibration into a parameter. Behold the latest prototype of the PolyPharma app:
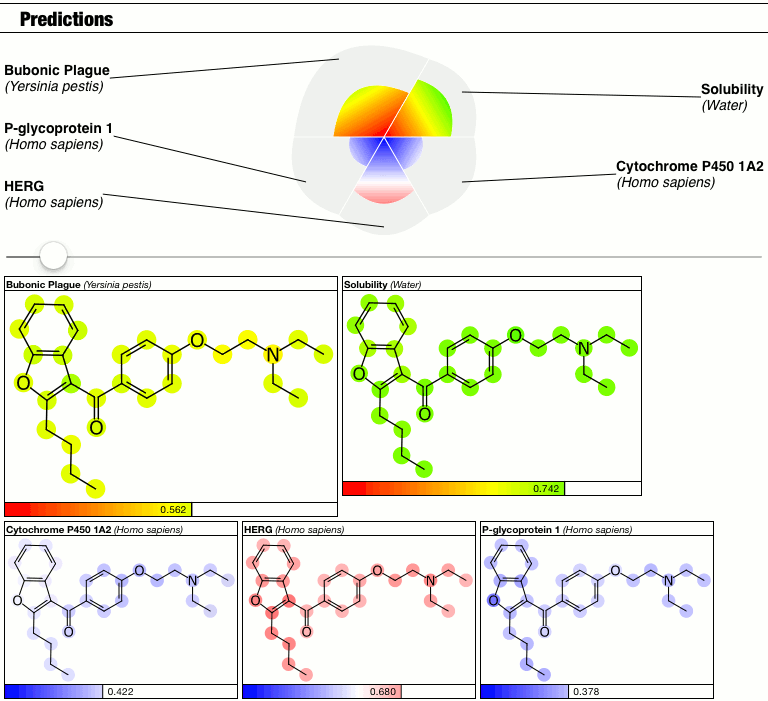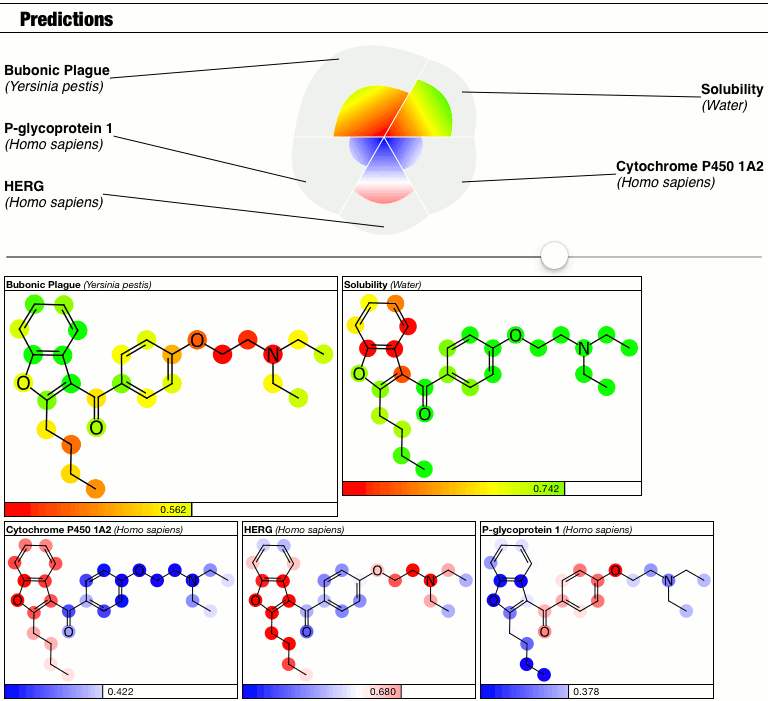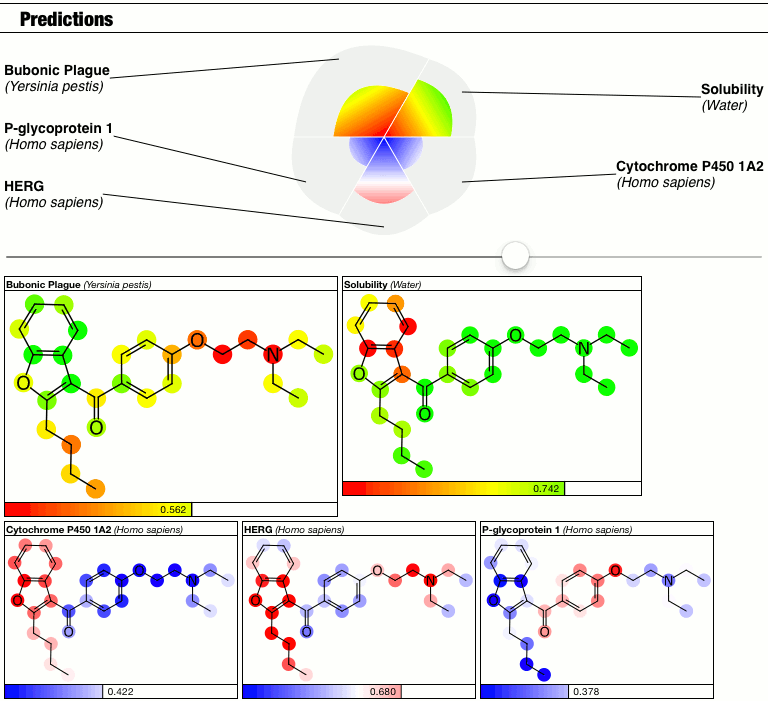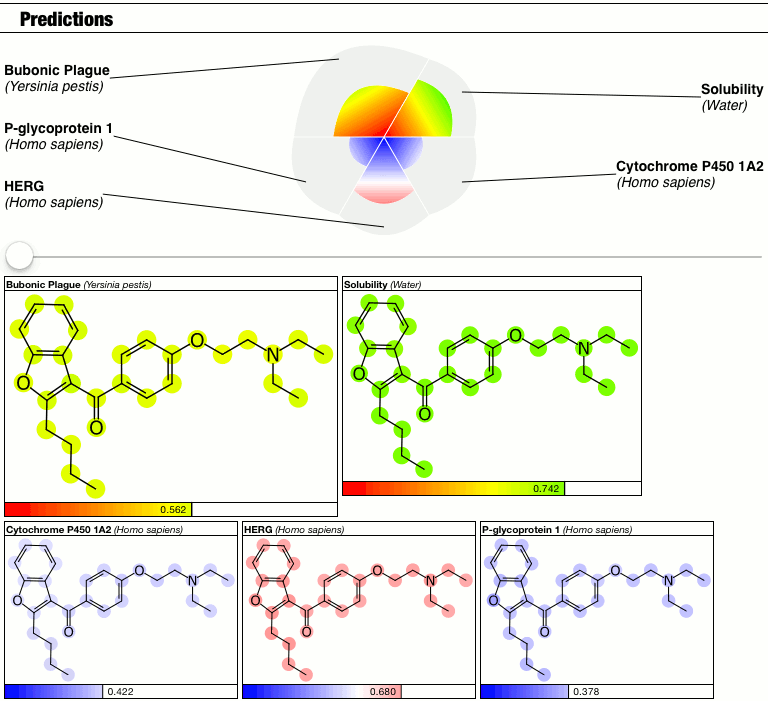
The top part of the predictions section now has a slider which controls the contrast, i.e. the calibration factor that brings attention to the fact that some atoms suggest greater contributions toward the overall goodness/badness of the Bayesian score. High contrast exaggerates the differences, low contrast blends them all in toward the average, which is the overall molecular prediction (which is calibrated).
Judgments about whether the good/bad regions are actually significant are now passed on to the scientific expertise of the operator, rather than solving this problem as part of the method.
But at least I admit it.

PolyPharma app is now live: http://itunes.apple.com/app/polypharma/id1025327772