 A newly available collection of antiviral structures from Chemical Abstracts has been made available, and has now been shoehorned into a model that can be used online to evaluate potential antiviral drug candidates for COVID-19. The tool can be found at https://molmatinf.com/covid19.
A newly available collection of antiviral structures from Chemical Abstracts has been made available, and has now been shoehorned into a model that can be used online to evaluate potential antiviral drug candidates for COVID-19. The tool can be found at https://molmatinf.com/covid19.
The third in this cheminformatics from quarantine series involves construction of a second model, which complements the original fragment binders. This was made possible due to the release of public data by Chemical Abstracts regarding structures of anti-viral compounds. The release description states that there are just under 50,000 structures of molecules which are either known antiviral drugs, or are structurally similar to those which are. At the time of writing I’m not exactly sure what they mean by this, but I would guess that it involved starting with specific structures curated as antiviral drugs, and then scooping up every other compound in the database that only differs by stereochemistry.
If you want to look at the data, it requires filling in an online form, but the approval process is immediate and automatic. The downloadable is a standard SDfile. I would recommend not distributing the raw content too freely, because it does come with a legal notice, which you should read.
The data itself has no information on which viral organisms are affected by each of the molecules, so the best use case would be to measure whether a hypothetical molecule is in the general vicinity of anti-viral drugs. At this early stage in the R&D pipeline for the pandemic, that may well be enough to filter out some dead-end leads.
The other issue is that the dataset has no negative results, which means that it is not ready to be turned into a structure-activity model. But there is always the option of resorting to the classic workaround: just take a large collection of diverse molecules, and assume that they are all inactive. Even though that claim is unlikely to be literally true, statistically speaking, it’s close enough.
When I’m looking for a representative database of compounds that have been used for biomedical purposes, I generally defer to the latest release of ChEMBL, because it is a sample of published attempts to achieve something of biomedical relevance. It is heavily skewed toward mainstream ideas about what a drug should look like, but it contains enough oddball chemicals to make a convincing case that it’s not completely biased. Academics try all kinds of strange things just in case, which is preferable to just downloading the output from a giant robotic synthesis screen.
To build the negative dataset (“inactives”), I put together a workflow to read in all 1,940,733 molecules from ChEMBL 26, and calculate maximum similarity to the CAS anti-viral selection. Any molecule that had a Tanimoto (ECFP6) coefficient above 0.5 was discarded, leaving 1,898,534 (97.83%) of the database.
The two datasets were combined and rolled into a Bayesian model. Initially I was a little bit worried about whether two million compounds would break my model building implementation, since the way it’s coded at the moment does require loading all the content into memory (because it needs to rebuild the model several times for cross validation and generation of the ROC curve). On a modern computer, though, it worked out just fine. Admittedly the output model size turned out to be over 100 MB when allowing the full spectrum of ECFP6 fingerprints (32 bits means up to 4 billion unique hash codes), so folding them into 16-bits turned out to be a more pragmatic option, given that the model will be loaded over the web.
The model can be downloaded directly from https://molmatinf.com/covid19 (see antiviral.bayesian). One note is that it has an artificially high ROC integral (>0.99) which is due to the circular reasoning that went into the data preparation: the presumed negatives were stripped of compounds very similar to the actives. This is one of many examples where model building metrics can be misleading: the high value merely means that the model has a very high ability to distinguish between two different types of molecules. The important question is whether that distinction is relevant, and that’s where scientific judgment comes in.
The model can be explored by drawing or pasting in molecules for prediction, e.g.
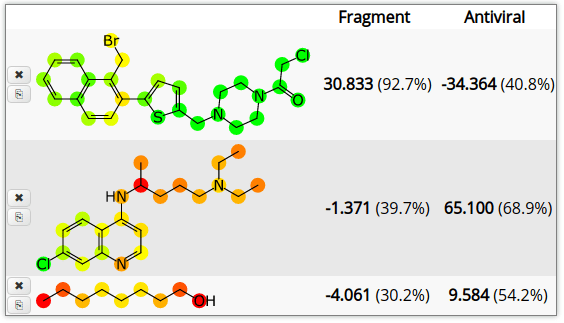
The top compound is one of the proposals (described in a recent article). It was constructed exclusively to score highly based on the fragment score, rather than being a ready-to-deploy antiviral drug. The second compound should be familiar to anyone who has been following the news. The third molecule is very non-druglike. As with the last update, the structures are colour-coded based on the atom contributions to the fragment model. The prediction score from the anti-viral model is added in a second column. The two models are based on very different concepts, so they are complementary. The fragment model is directly relevant since it is based on experimental data for this actual disease, whereas the anti-viral model encodes structure activity information about what makes an actual drug, which is important too.
Hopefully in the next days and weeks there will be many more datasets coming online which will allow us to do more and better modelling. Until then, there are various creative ways to make the best of what we have.
