 The latest experimental feature of the BioAssay Express project involves taking all of the curated assays (3500 so far) and their corresponding compounds from PubChem (hundreds of thousands of unique structures) and feeding them all into one giant Bayesian model. Rather than the usual approach of modelling compound ⟹ activity separately for each assay, this approach takes advantage of the fact that the semantic annotations that describe the assay can be used like fingerprints. The model is therefore compound + assay ⟹ activity, which means that it is possible to lump together everything that is known about screening compounds against assay protocols.
The latest experimental feature of the BioAssay Express project involves taking all of the curated assays (3500 so far) and their corresponding compounds from PubChem (hundreds of thousands of unique structures) and feeding them all into one giant Bayesian model. Rather than the usual approach of modelling compound ⟹ activity separately for each assay, this approach takes advantage of the fact that the semantic annotations that describe the assay can be used like fingerprints. The model is therefore compound + assay ⟹ activity, which means that it is possible to lump together everything that is known about screening compounds against assay protocols.
Whether this is actually a good idea or not is to be determined: this will be the next step for the project, but right at this moment in time, the machinery for asking that question is freshly baked.
The modelling technology of choice for the moment is the trustworthy Bayesian variant that has been serving drug discovery effectively for awhile: chemical structures are converted into fingerprint bitstrings using so-called circular fingerprints, and these are turned into a model by adding up the ratios of actives vs. inactives for each fingerprint. That is essentially all there is to a Bayesian model, making it very fast and easy to create (although the cross validation and calibration metrics take a bit more effort). Bayesian models have a few other advantages besides being easy to implement and fast to execute: they are also relatively resistant to overtraining, and are very easy to interpret (which can be demonstrated graphically with structure fragments).
Back to the BioAssay Express: the core idea of this project is to describe assay protocols by using semantic web terms rather than text. Each annotation is a URI, and it not only has a clear and well defined meaning that has been pre-established for interpretation by either machines or humans, but it also has an implicit hierarchy that brings additional value.
For example, assigning the annotation of:
[http://www.bioassayontology.org/bao#BAX_0000010,
http://purl.obolibrary.org/obo/DOID_401%5D
… probably doesn’t leap out at you and explain itself, but it can be alternatively explained as:
property: applies to disease
value: disease
➥ disease by infectious agent
➥ bacterial infectious disease
➥ primary bacterial infectious disease
➥ tuberculosis
➥ multidrug-resistant tuberculosis
Each of these items in the annotation trail can be treated like a fingerprint, i.e. 6 of them from the nested hierarchy for this annotation alone. Another assay protocol that is setup to screen for multidrug-resistant tuberculosis would have 6 out of 6 fingerprints in common, whereas an assay that screens against some other kind of primary bacterial infectious disease would share 4 out of 6.
In this way all of the assays that are being thrown into the model can be given a series of fingerprints for their various annotations, which they either possess, or do not. This is the appropriate form for feeding into the question half of a Bayesian model. But we still need to plug something in for the answer, since an assay protocol isn’t generally referred to as active or inactive.
Enter the compounds: PubChem conveniently assembles a list of chemical structures for each assay, along with an executive decision on behalf of the data submitter as to whether the compound was considered active against that screen. Which means that if we expand out all of the assays ✖️ compounds, we get the right materials, something like:
{assay1},{compound1} = {compound1 active?}
{assay1},{compound2} = {compound2 active?}
…
{assay2},{compound3} = {compound3 active?}
{assay2},{compound4} = {compound4 active?}
… etc …
Each compound brings with it a collection of structure-derived fingerprints (ECFP6) and assay-derived fingerprints.
This model building formalism has now been implemented in the heart of the BioAssay Express service, and is one of the many background tasks: every time the data changes, the models are rebuilt. The models themselves are prescribed ahead of time by specifying query terms, which are rederived to select the assays for each model building exercise; there are currently three of them defined – modelling everything at once, and separating into primary vs. secondary assay screens:
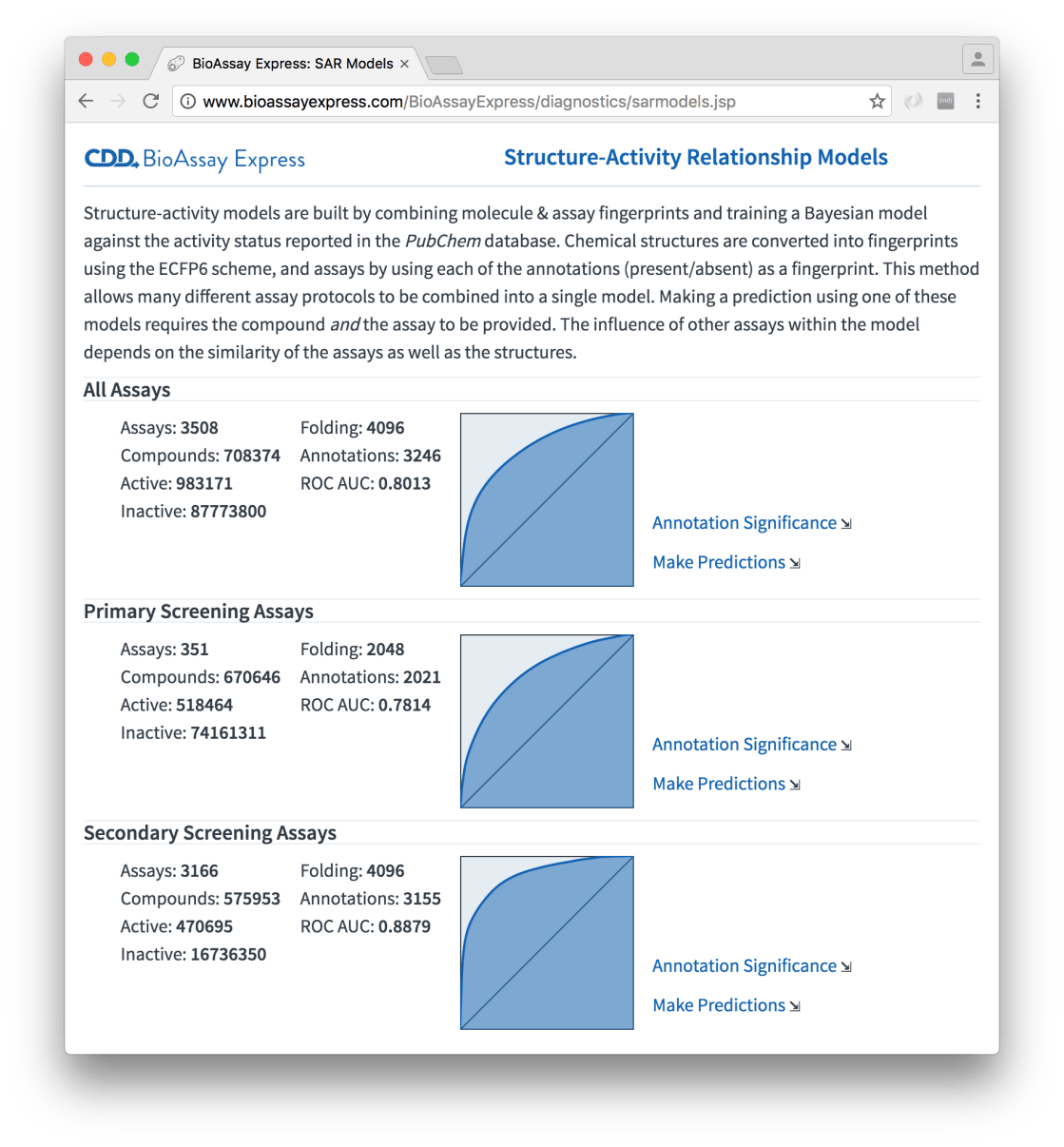
The summary shows preliminary information about each model, and as you can see, the ROC curves are fairly good, as far as large models of relatively raw structure-activity data go. For each model there are two important links: the first one is Annotation Significance:
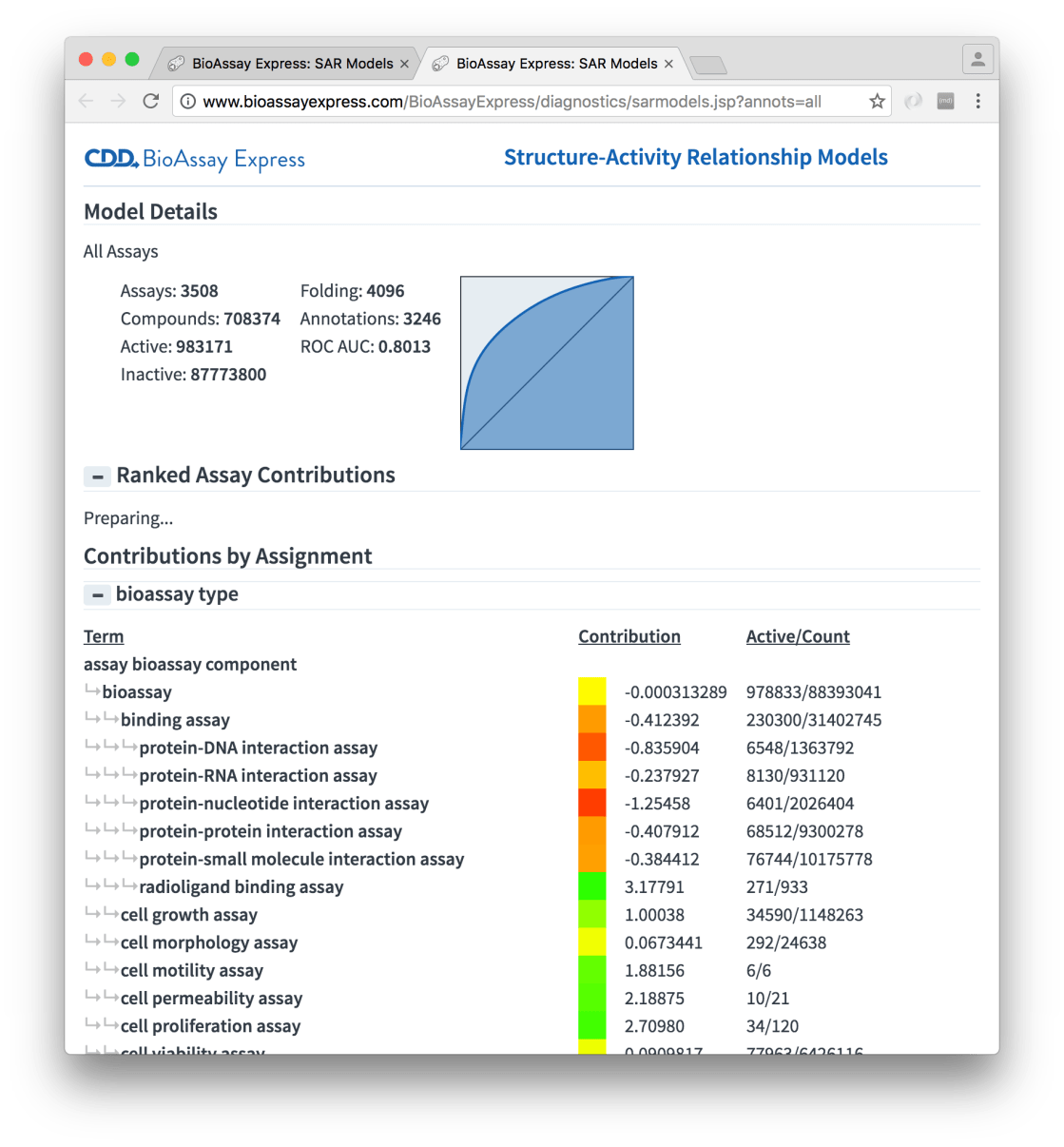
This report is more than just an idle curiosity: it provides two views into the assay annotation terms and their contributions to the model. Each of the assignment properties (in the above snapshot, bioassay type is partially listed) is shown in a hierarchy, with all of the terms that got included in the model being represented. Each of these terms have a contribution to the underlying Bayesian model, which is composed from the portion of actives with that fingerprint. This is summarised using heatmap colour-coding, i.e. green blocks indicate that the term is frequently found in actives, while red means that this is much less so, and yellow in the middle somewhere.
The other detail page for a model is Make Predictions, which offers to do exactly what the name suggests:
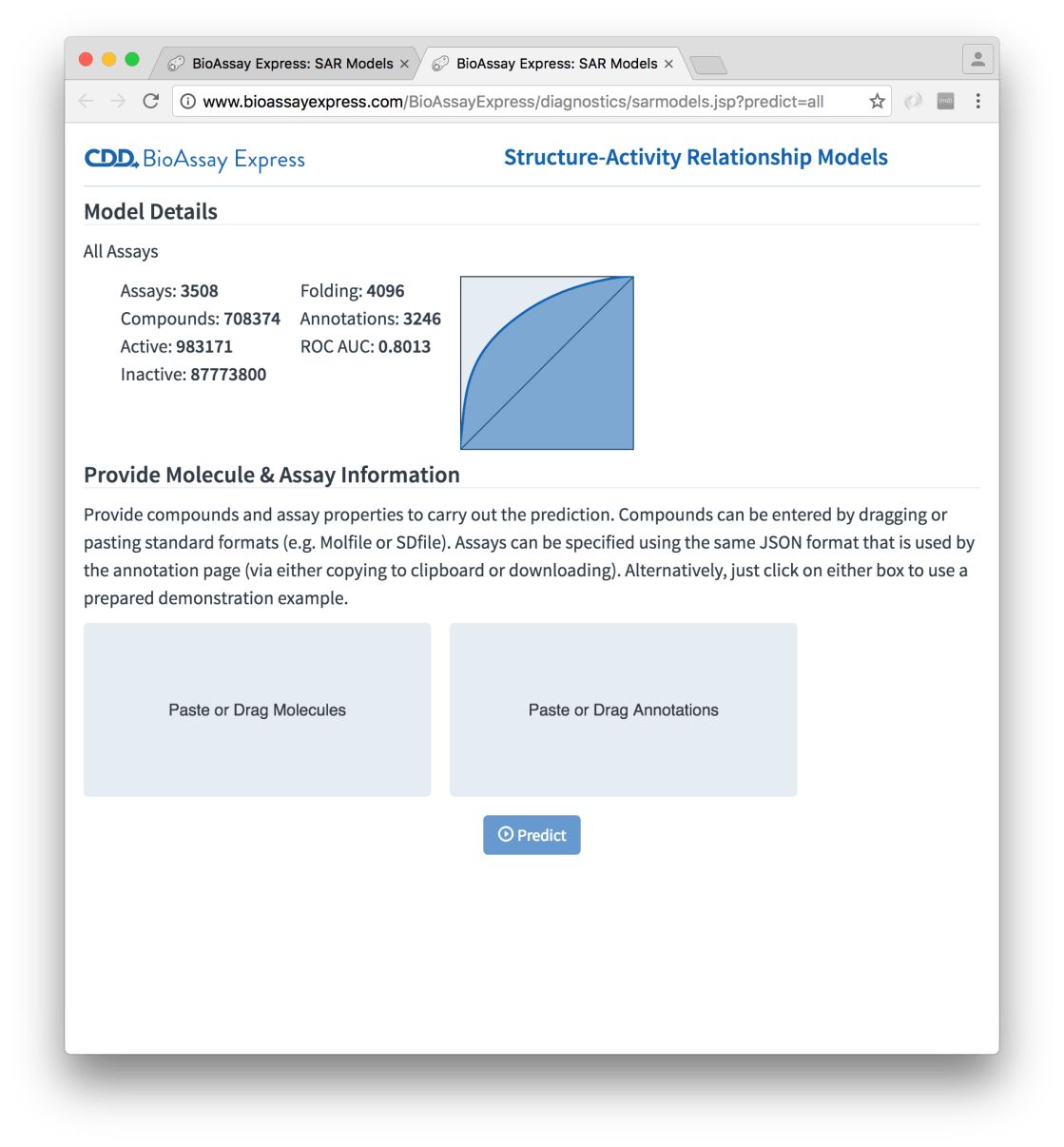
The page is initially dominated by two rectangles, which invite you to provide molecules and annotations respectively. Pasting a molecule (e.g. a Molfile on the clipboard) or dragging a collection of molecules (e.g. an SDfile from the file manager) into the molecule box will parse and extract the structures. Similarly for annotations: these can be produced using the Assign page (the web UI allows both copying to clipboard and saving as a separate file). Or if you’re in a hurry, just click on each of the rectangles, to get a prefabricated example. With some plausible inputs, the setup looks like:
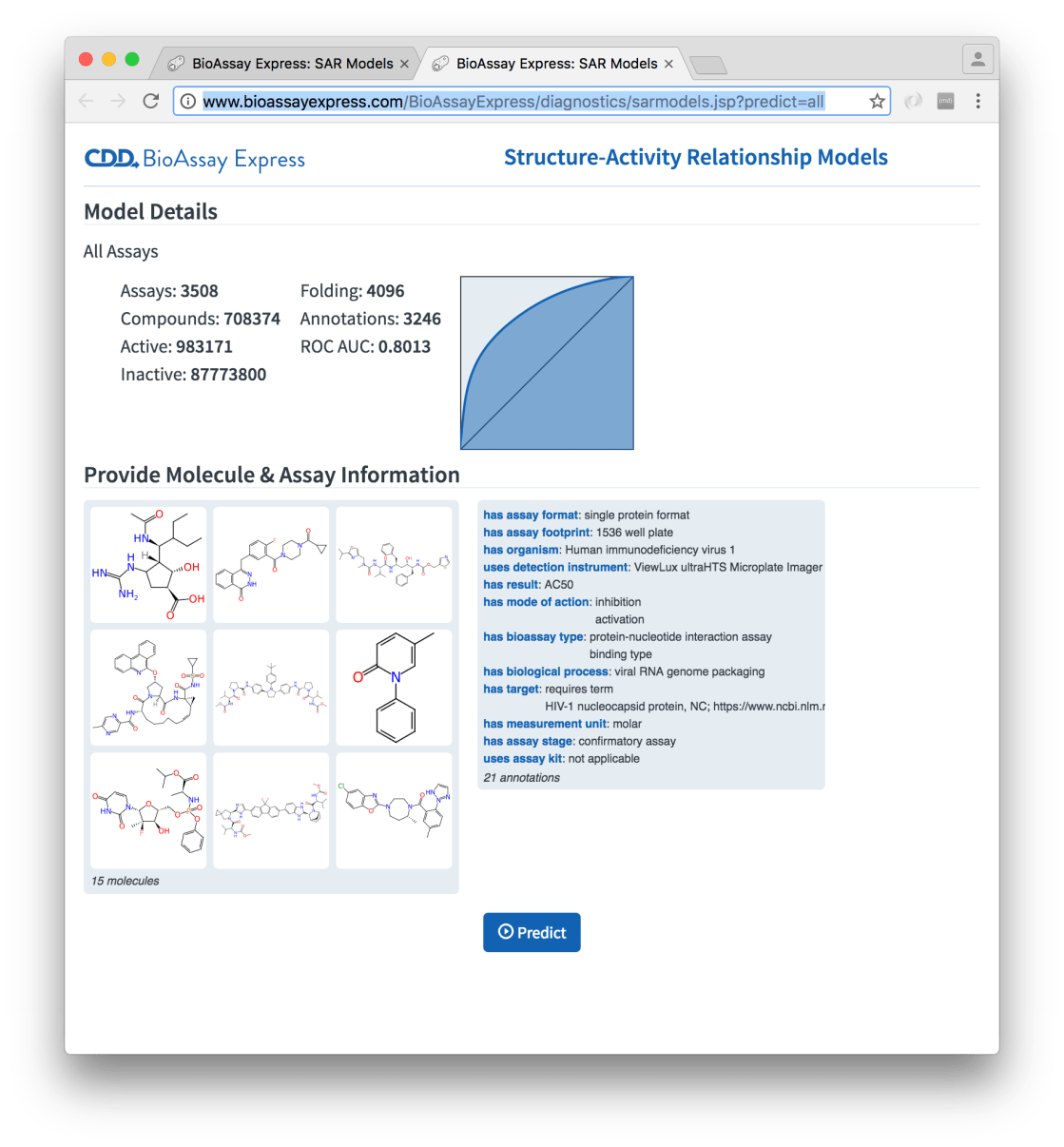
The left hand panel now shows a preview of the molecules that were dragged onto the page (which may look familiar to anyone who has been following newly released drugs), and likewise the right hand panel shows a summary of the assay description that will be used for the prediction. In this case the annotations are taken from an actual assay protocol, but this does not have to be the case: it could be formulated as a hypothetical protocol that has yet to be run.
Pressing the Predict button uses the model to make an activity estimate for each of the compounds, and these are displayed underneath:
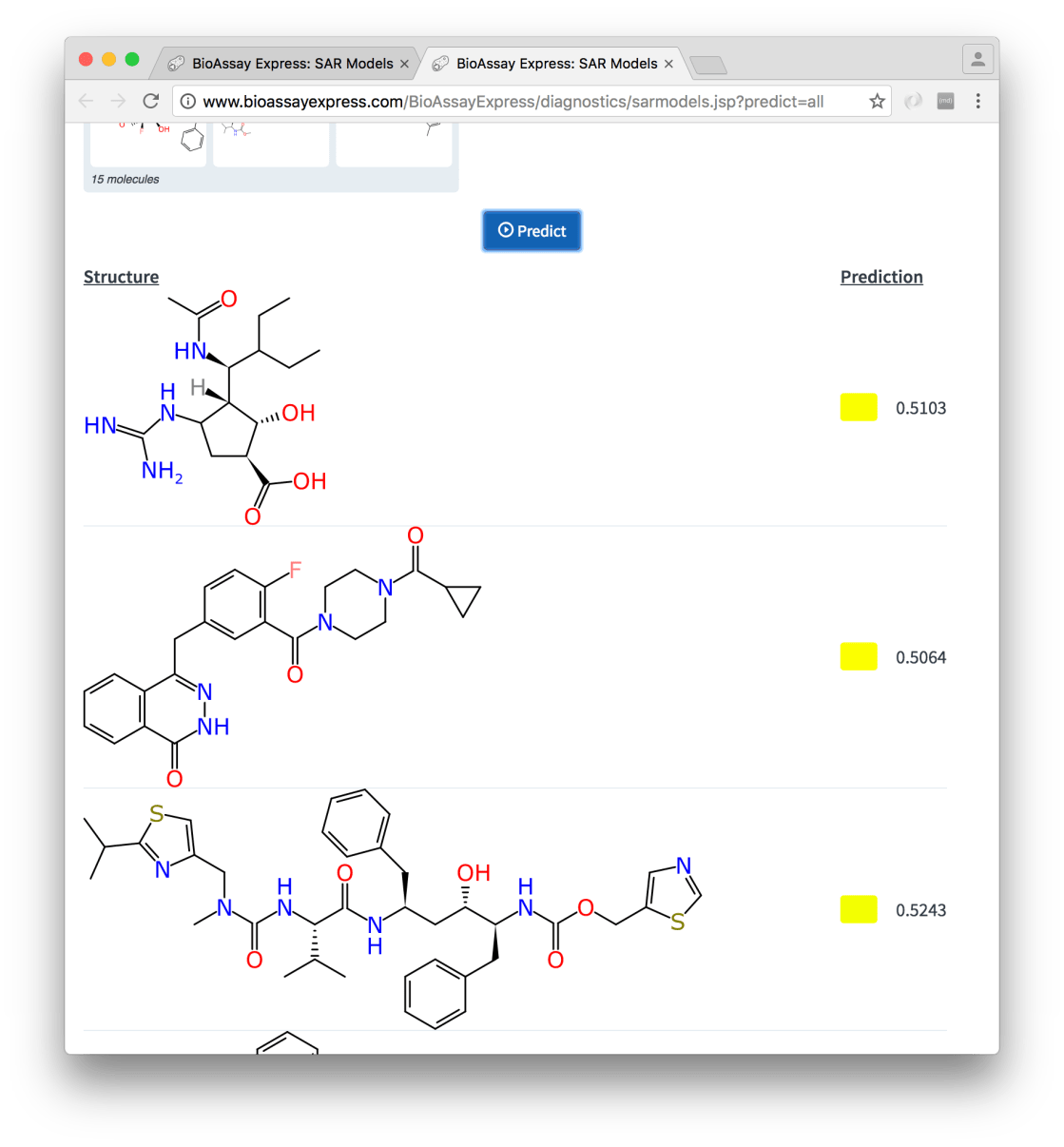
Among other things, there is some technical novelty behind the scenes: the application of the mixed compound/assay Bayesian model is done entirely on the client side, which is made possible by the fact that the structure fingerprints (ECFP6) are calculated on the client side, by using a very recent port of our open source implementation to TypeScript. The molecular graphics are also prepared and displayed without any help from the server.
From the drug discovery point of view, this yet-to-be-validated approach to structure-activity prediction is interesting because all of the assays are mixed together into the same model, which means that there will be spillover from assays that are not related to the target that you are interested in. Normally you would not be inclined to build a structure-activity model by mixing data from two completely different screens: but if you did, you might find certain trends, for example, certain structural features always seem to light up most high throughput screens regardless of the target; or certain detection methods are especially harsh or forgiving for certain structural characteristics; or for the same disease and similar compounds, a certain assay kit or cell line results in a higher or lower hit rate.
The ability to leverage similar assays to enrich prediction data is another potential benefit. For example, running models for an antibacterial study against a new target may well produce useful results because it can selectively splice in positive/negative predictivity based on data acquired for related targets using similar assay protocol setups. The alternative would be to skip the modelling step altogether if negligible data was available, or perhaps make several predictions against models for similar targets, and try to interpret the results individually.
While I’m completely convinced that the idea of mixing structure and assay descriptors together (which is only possible now that we have these semantic annotations) is an approach that will lead to useful results, it remains to be seen whether the naïve Bayesian method is up to the task of exploiting this mixture. The simple Bayesian method lacks any way to internally correlate priors or identify localised subgroups, and so it may be necessary to look for much more advanced techniques such as deep neural networks to come up with really impressive predictions. Or maybe not: perhaps enough trends will be captured by the trivially simple Bayesian formalism to be useful. And of course we do have the option of partitioning into as many model subsets as necessary (i.e. fixing certain properties in place, while allowing others to define the model).
Watch this space: when we have promising results, we’ll definitely want people to know about it.
