 The OS X Molecular DataSheet (XMDS) desktop app is now ready for beta testing. And by beta testing, I mean the minimum viable product is done and ready to be used for actual cheminformatics tasks. You can sign up anytime: all it takes is an email (info@molmatinf.com), a Mac with Yosemite-or-later (v10.10), and a Dropbox account. And you get to keep using the app for as long as you want, even after the beta testing programme is wrapped up. Continue reading
The OS X Molecular DataSheet (XMDS) desktop app is now ready for beta testing. And by beta testing, I mean the minimum viable product is done and ready to be used for actual cheminformatics tasks. You can sign up anytime: all it takes is an email (info@molmatinf.com), a Mac with Yosemite-or-later (v10.10), and a Dropbox account. And you get to keep using the app for as long as you want, even after the beta testing programme is wrapped up. Continue reading
Literature how-to for structure:activity Bayesian models (and open source)
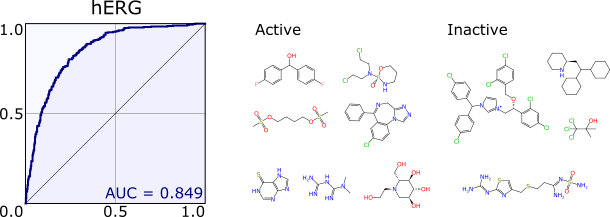 A two-pack of publications in Journal of Chemical Information and Modeling is now available: Bayesian the first, and Bayesian the second. Both papers are open access, so by all means go read them instead of this blog post. The first paper details the implementation of a variation of the classic naive Bayesian method that is suitable for use with structure-derived fingerprints such as ECFP6 and FCFP6. The text goes into some detail about how it is implemented, to the point of including pseudocode, which complements the fact that the source code is available as part of the Chemical Development Kit (CDK), conveniently and concisely coded up in a single source file. The intention is quite unashamedly to tell you everything you need to know to build the algorithm from scratch, should you be so inclined; and if not, to understand every little detail about how the open source software works. The second paper goes into some more detail about how to use this kind of (“Laplacian-modified”) Bayesian model, including a calibration method, and an extensive study carried out by extracting thousands of model-ready datasets from the ChEMBL database. Continue reading
A two-pack of publications in Journal of Chemical Information and Modeling is now available: Bayesian the first, and Bayesian the second. Both papers are open access, so by all means go read them instead of this blog post. The first paper details the implementation of a variation of the classic naive Bayesian method that is suitable for use with structure-derived fingerprints such as ECFP6 and FCFP6. The text goes into some detail about how it is implemented, to the point of including pseudocode, which complements the fact that the source code is available as part of the Chemical Development Kit (CDK), conveniently and concisely coded up in a single source file. The intention is quite unashamedly to tell you everything you need to know to build the algorithm from scratch, should you be so inclined; and if not, to understand every little detail about how the open source software works. The second paper goes into some more detail about how to use this kind of (“Laplacian-modified”) Bayesian model, including a calibration method, and an extensive study carried out by extracting thousands of model-ready datasets from the ChEMBL database. Continue reading
Honeycomb clustering in Approved Drugs app: sneak preview
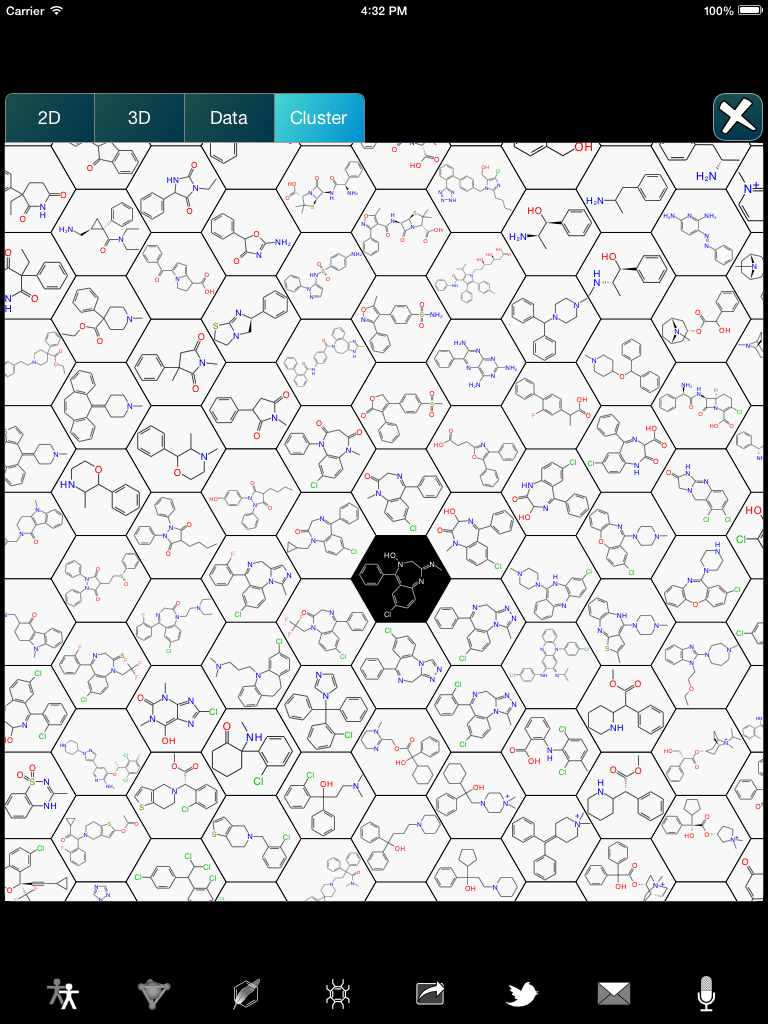 Work is currently underway on a novelty feature that will first be exposed within the Approved Drugs app: honeycomb clustering, which is a greedy visualisation technique that is remarkably effective for examining how a particular chemical structure relates to a collection of compounds. Continue reading
Work is currently underway on a novelty feature that will first be exposed within the Approved Drugs app: honeycomb clustering, which is a greedy visualisation technique that is remarkably effective for examining how a particular chemical structure relates to a collection of compounds. Continue reading
XMDS and dragging content into other Mac apps
 One of the things that one must become accustomed to when using a Mac regularly is that drag’n’drop is actually an effective way to get real work done. Two main use case scenarios are considered: getting graphics images of molecules into presentation tools (e.g. Pages or Keynote), and getting raw data into another cheminformatics tool (e.g. uploading an SDfile to a browser-based application). Both of these are now functioning in the pre-alpha version of XMDS (the OS X Molecular DataSheet), which essentially brings it to demo-ready status. Continue reading
One of the things that one must become accustomed to when using a Mac regularly is that drag’n’drop is actually an effective way to get real work done. Two main use case scenarios are considered: getting graphics images of molecules into presentation tools (e.g. Pages or Keynote), and getting raw data into another cheminformatics tool (e.g. uploading an SDfile to a browser-based application). Both of these are now functioning in the pre-alpha version of XMDS (the OS X Molecular DataSheet), which essentially brings it to demo-ready status. Continue reading
Summer conference schedule
I’ve been out of the spotlight for awhile as far as scientific conferences go, but this summer-of-2015 it’s all back in form, with three in a month long window:
- 12-14 July: Green Chemistry & Engineering (Washington D.C.) – “Green chemistry in chemical reactions: Informatics by design“
- 19-24 July: Gordon Research Conference/Computer Aided Drug Design (Vermont) – “CADD of the future: using tablets and phones for drug discovery, and learning from the parallel universe of consumer electronics” [poster]
- 16-20 August: American Chemical Society (Boston) – “Compact models for compact devices: Visualisation of SAR data using mobile apps” and “Anatomy of a chemical reaction: Dissection by machine learning algorithms”
If you’re going to be attending any of these, perhaps I will see you there. If not, the materials will be going up on slideshare shortly thereafter…
Medicinal Chemistry Toolkit app, now with structures and calculations
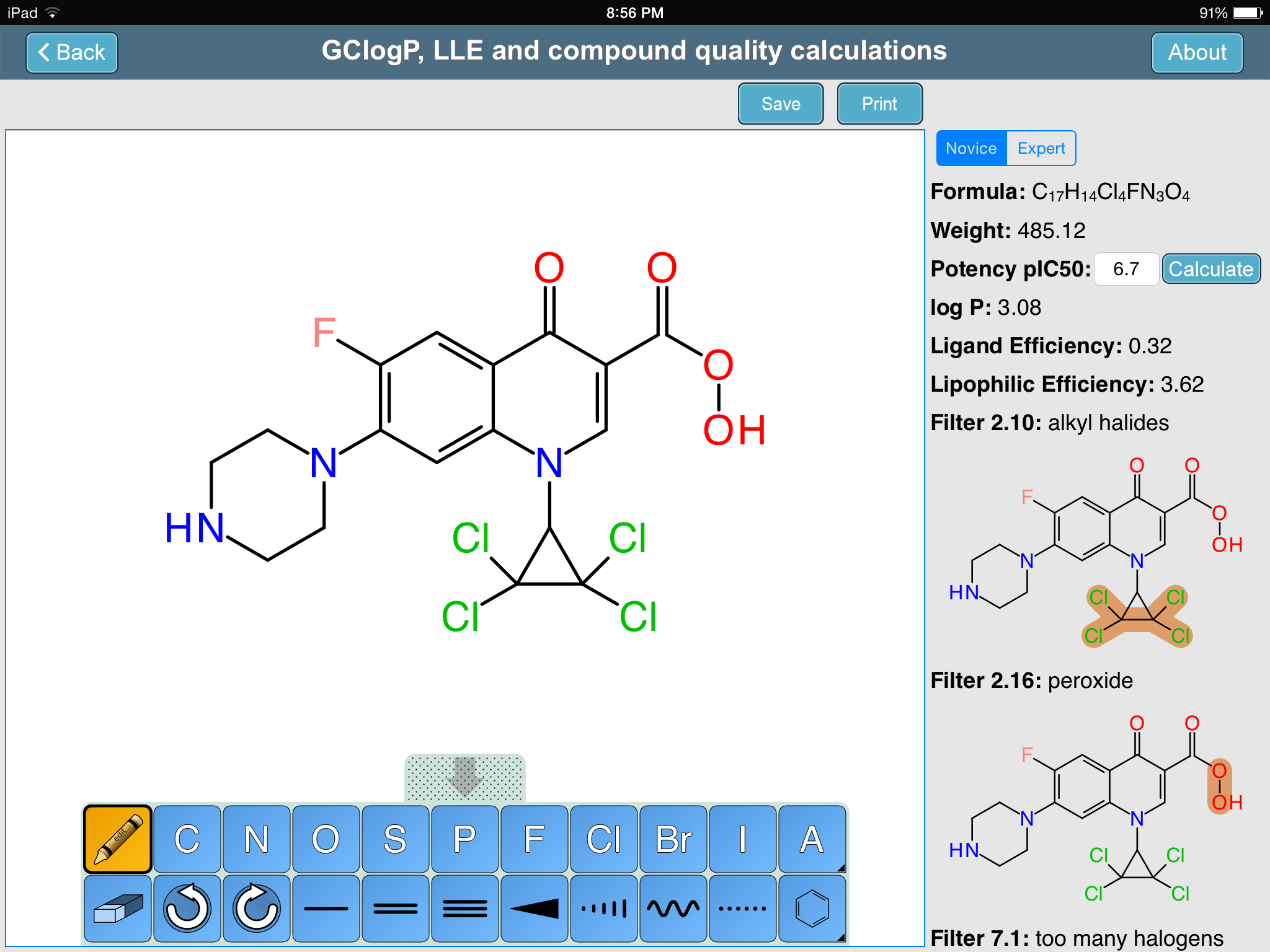
The Royal Society’s Medicinal Chemistry Toolkit app has been up on the AppStore since late last year, but a couple days ago it got updated with some major new functionality: an interactive tool that allows a structure to be drawn, and various properties to be calculated from it. If you can’t guess who supplied the sketcher and the structure-derived calculations, you’re probably not a regular on this blog. Continue reading
Ergonomic molecule editing, and praise for Mac-style dialogs
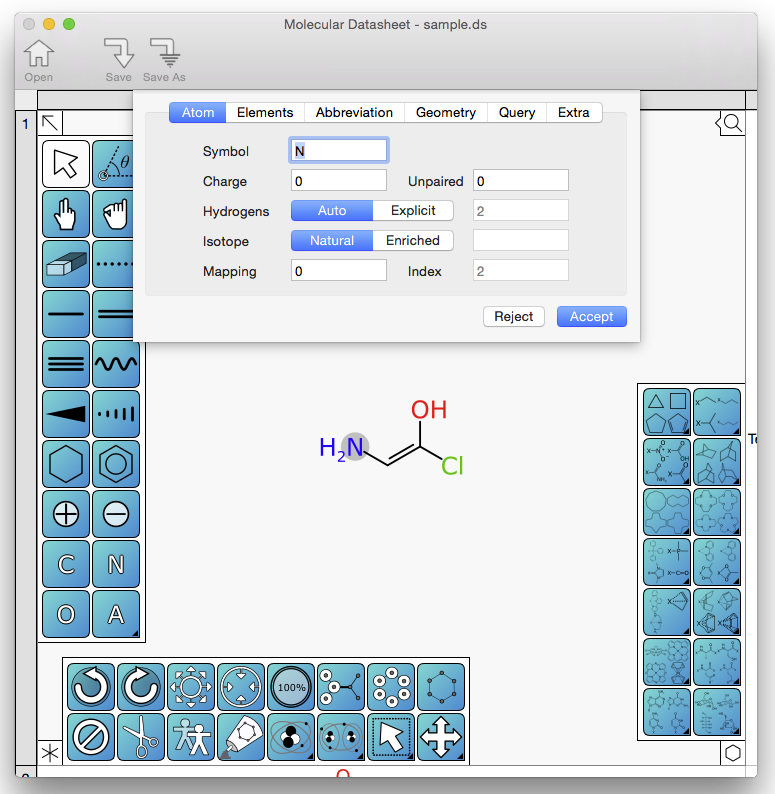 Normally I wouldn’t consider a trivial dialog box to be worthy of a blog post, but it’s as good a time as any for a progress update on XMDS. The sketching interface for editing molecular structures is now somewhat feature complete. There are a few bits to come back to, but in general the methods for adding, deleting, editing and aligning atoms and bonds are available in a highly redundant way. By redundant, I mean that in a good way. The editor is based on a confluence of methodologies that I’ve built out in various products over the years: (1) the conventional way of drawing molecules with a painting-style toolbox (see SketchEl); (2) the “drawing primitives” designed for precise editing when user interaction is expensive, motivated by mobile devices with tiny screens; and (3) keyboard shortcuts, designed so that an expert can draw molecules incredibly quickly without having to reach for the mouse, battle-tested with the BlackBerry version of MMDS back in 2010, when touchscreens were not yet ubiquitous. Continue reading
Normally I wouldn’t consider a trivial dialog box to be worthy of a blog post, but it’s as good a time as any for a progress update on XMDS. The sketching interface for editing molecular structures is now somewhat feature complete. There are a few bits to come back to, but in general the methods for adding, deleting, editing and aligning atoms and bonds are available in a highly redundant way. By redundant, I mean that in a good way. The editor is based on a confluence of methodologies that I’ve built out in various products over the years: (1) the conventional way of drawing molecules with a painting-style toolbox (see SketchEl); (2) the “drawing primitives” designed for precise editing when user interaction is expensive, motivated by mobile devices with tiny screens; and (3) keyboard shortcuts, designed so that an expert can draw molecules incredibly quickly without having to reach for the mouse, battle-tested with the BlackBerry version of MMDS back in 2010, when touchscreens were not yet ubiquitous. Continue reading
Swift gets a bit swifter: version 1.2 first impressions
Xcode 6.3 appeared in my update tray this morning, and since it contains Swift 1.2, I installed it right away. The first thing I noticed, after fixing about 200 compiler errors due to minor changes in language syntax, was that the work-in-progress XMDS app all of a sudden got really snappy. Rather than feeling like using a computer that’s 10 years obsolete, algorithms that were borderline rate limiting running in the main UI thread just happen like they ought to. As a reality check, I re-ran the horrendously underperforming algorithm that I complained about awhile back, and rather than taking 320 seconds to calculate 7 log P values, it now gets the job done in 30 seconds. That comparison is with standard compiler options. The alternate target with all the optimisation settings dialled up actually crashes the Swift compiler, so no metrics for that.
Nonetheless, 10x improvement in a scenario that’s relevant to cheminformatics, and a qualitative observation that this seems to be representative for GUI tasks, is a big deal. I’m sure there’s a lot more fat to trim over the coming years, since the Swift syntax is designed in a way that allows the compiler to do some pretty hardcore optimisation. Getting the practical implementation levelled up to “adequate” is a good start!
XMDS takes another step: copying molecule into CDD Vault
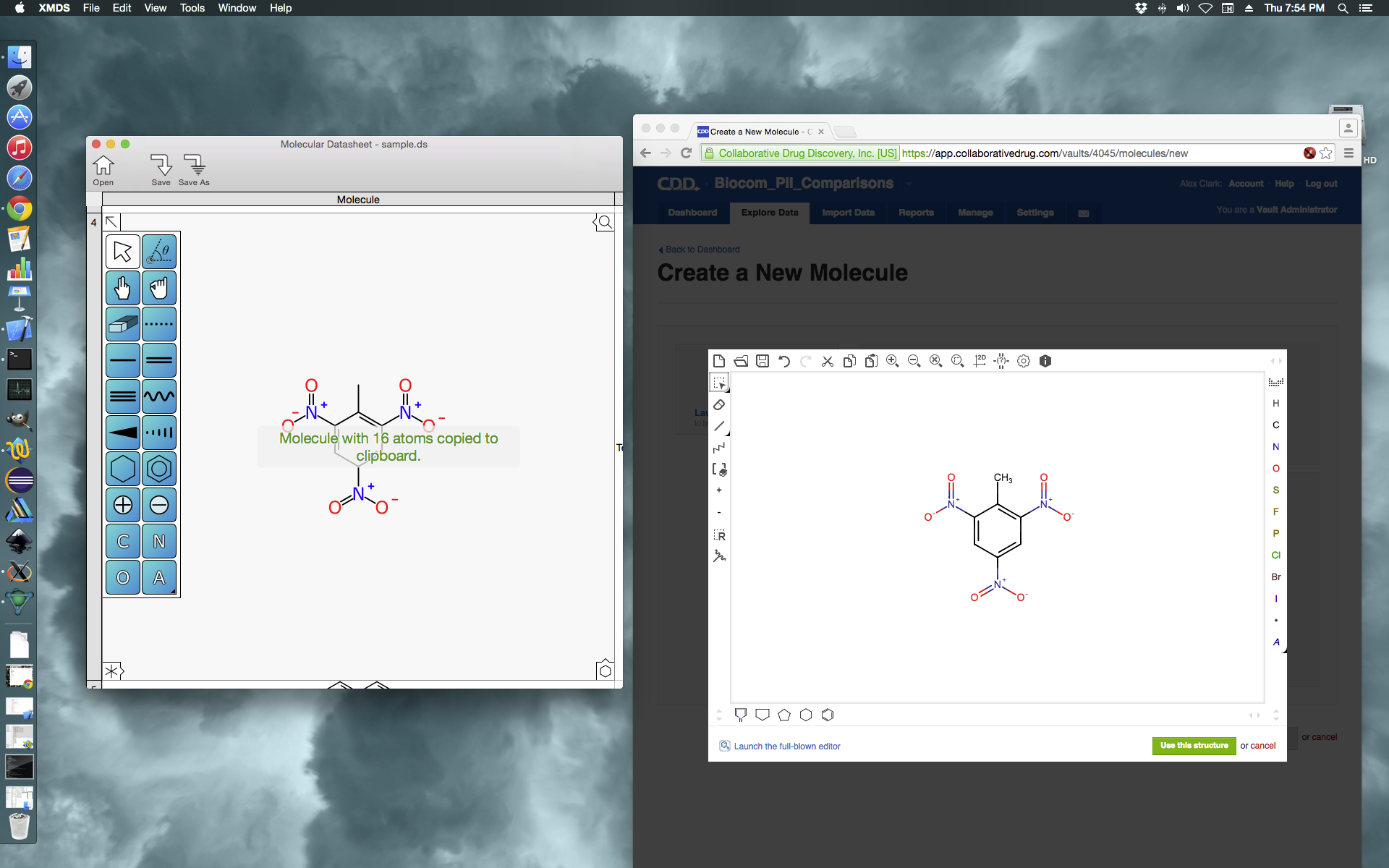 It may be a slightly arbitrary milestone, but the Mac app-in-progress XMDS has performed the first instance of a deed that might be argued as being useful. Most of the sketching functionality is now operational, which combines pretty much all of the drawing capabilities of the MMDS app and the SketchEl desktop structure drawing tool, as well as a few new ideas. The feature that was added today is clipboard interoperability, which means that structures can be used elsewhere. In this screenshot, the structure has been pasted into an instance of CDD Vault in the browser, in draw-new-molecule mode.
It may be a slightly arbitrary milestone, but the Mac app-in-progress XMDS has performed the first instance of a deed that might be argued as being useful. Most of the sketching functionality is now operational, which combines pretty much all of the drawing capabilities of the MMDS app and the SketchEl desktop structure drawing tool, as well as a few new ideas. The feature that was added today is clipboard interoperability, which means that structures can be used elsewhere. In this screenshot, the structure has been pasted into an instance of CDD Vault in the browser, in draw-new-molecule mode.
A rant about data quality: machines first, humans second…
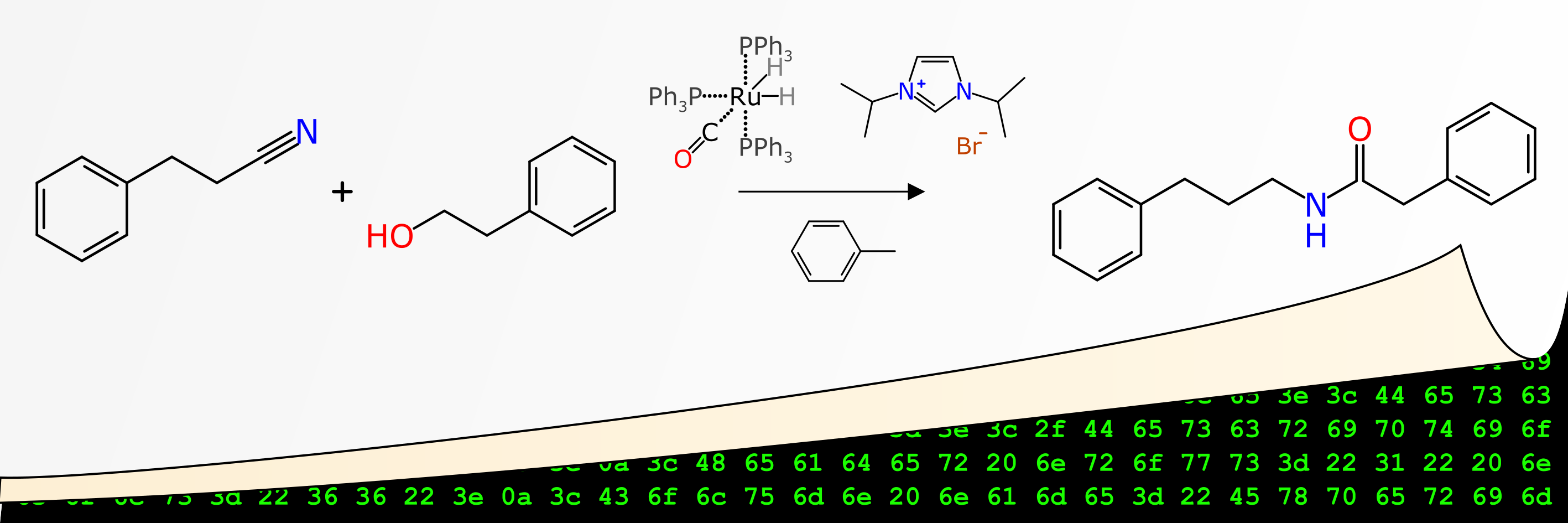 Recently one of my papers emerged through the publication system of Journal of Cheminformatics, entitled “Machines first, humans second: on the importance of algorithmic interpretation of open chemistry data“, co-authored with Antony Williams and Sean Ekins, and incorporated into the JC Bradley Memorial Issue. Spoiler alert: the paper is about how if you’re publishing open lab notebook data without adhering to rigorously defined standards for machine readability, then you’re mostly wasting your time, and arguably making the open data situation even worse than it already is. The tone of the article is a bit less polite than I normally try to be, so fair warning, but it’s all for a good cause.
Recently one of my papers emerged through the publication system of Journal of Cheminformatics, entitled “Machines first, humans second: on the importance of algorithmic interpretation of open chemistry data“, co-authored with Antony Williams and Sean Ekins, and incorporated into the JC Bradley Memorial Issue. Spoiler alert: the paper is about how if you’re publishing open lab notebook data without adhering to rigorously defined standards for machine readability, then you’re mostly wasting your time, and arguably making the open data situation even worse than it already is. The tone of the article is a bit less polite than I normally try to be, so fair warning, but it’s all for a good cause.
